Tackling the Antibiotic Resistance Caused by Class A β-Lactamases through the Use of β-Lactamase Inhibitory Protein


Abstract
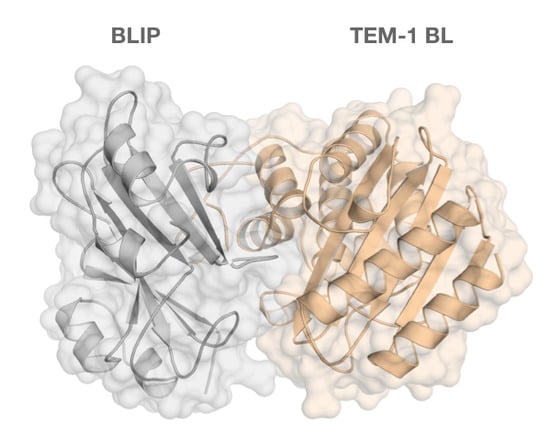
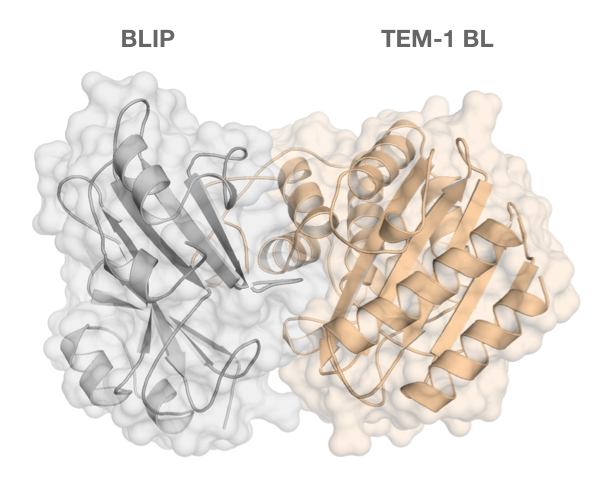
:
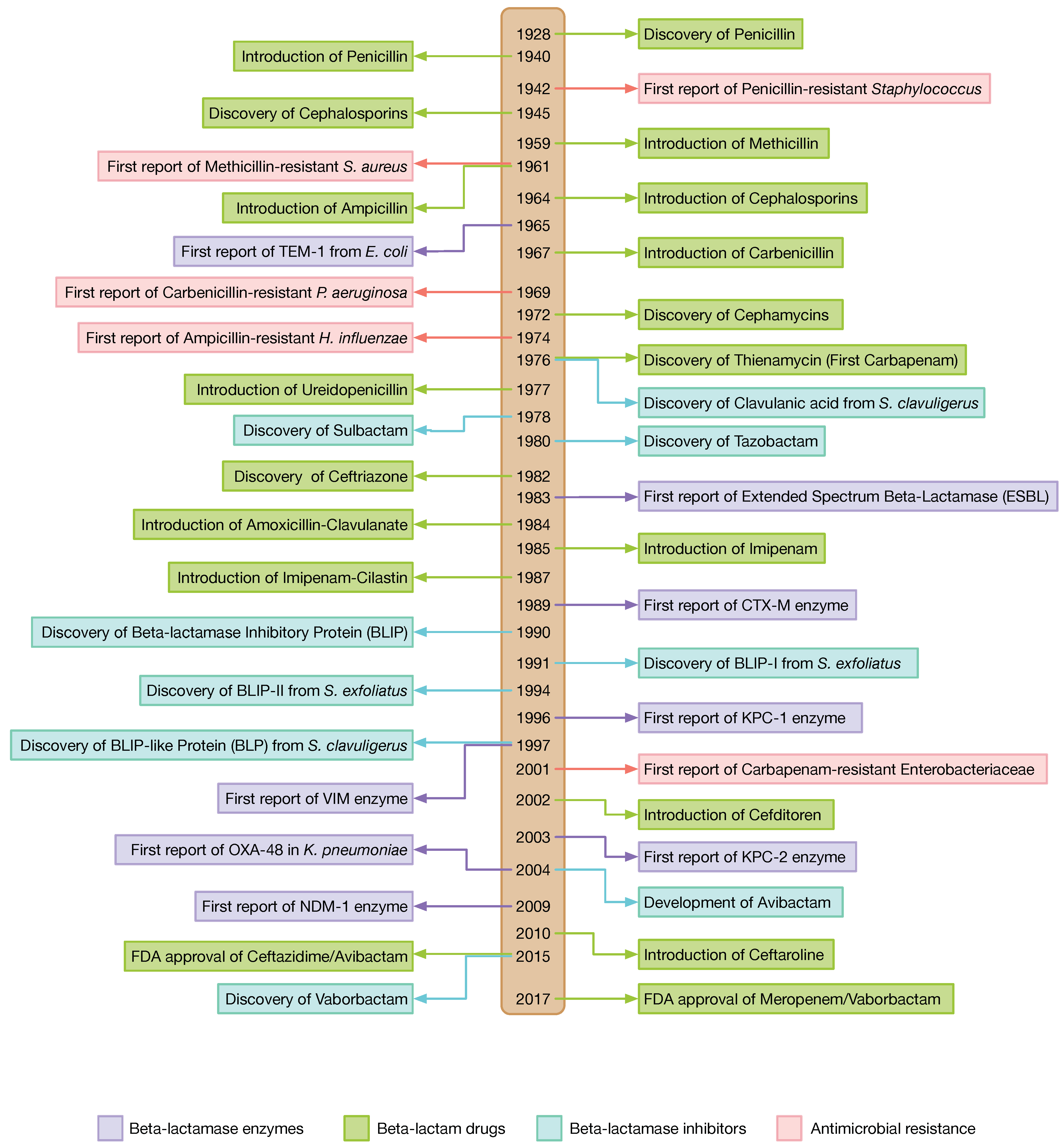
1. Introduction
2. -Lactamases
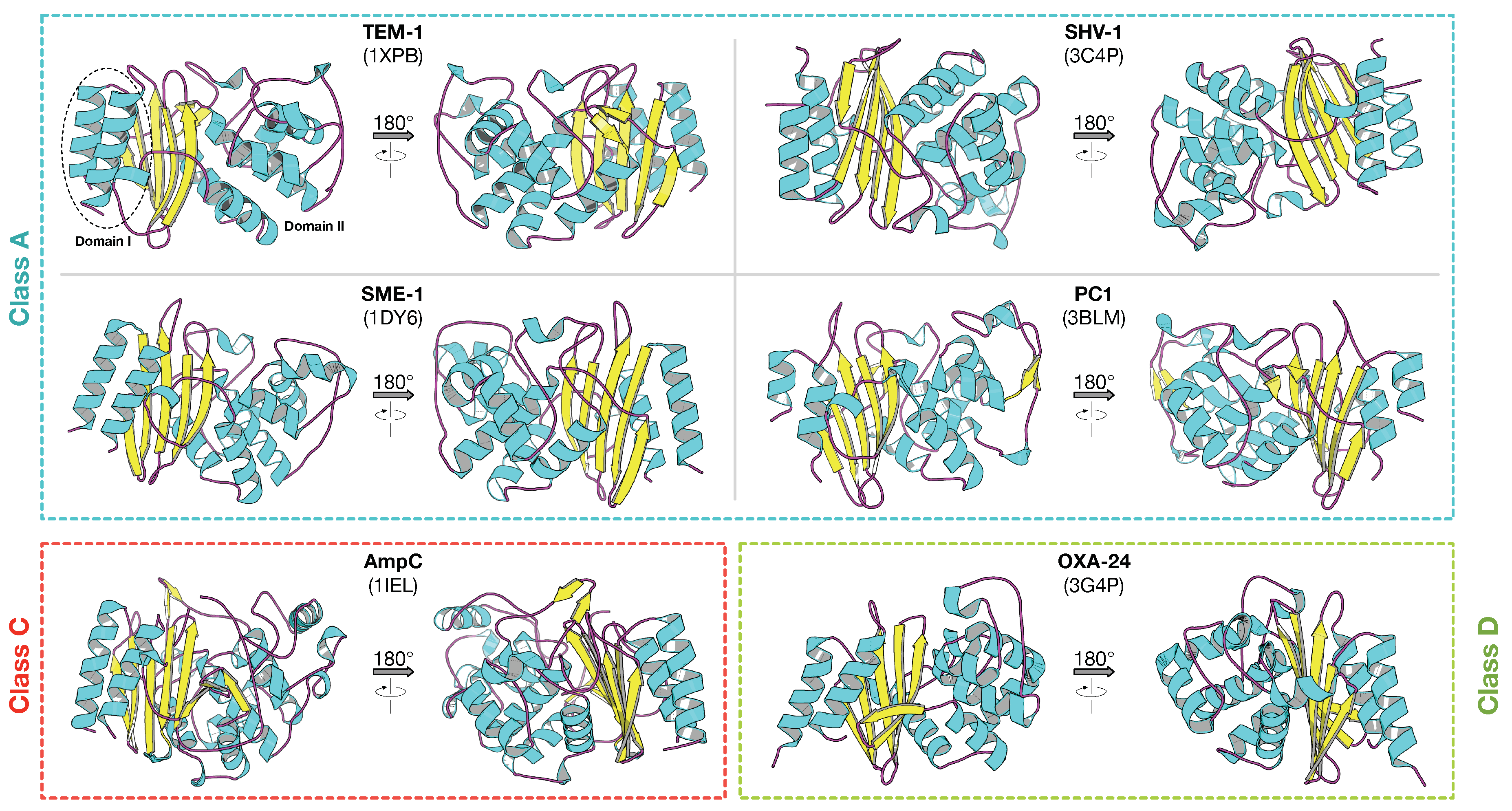
2.1. Class A -Lactamase
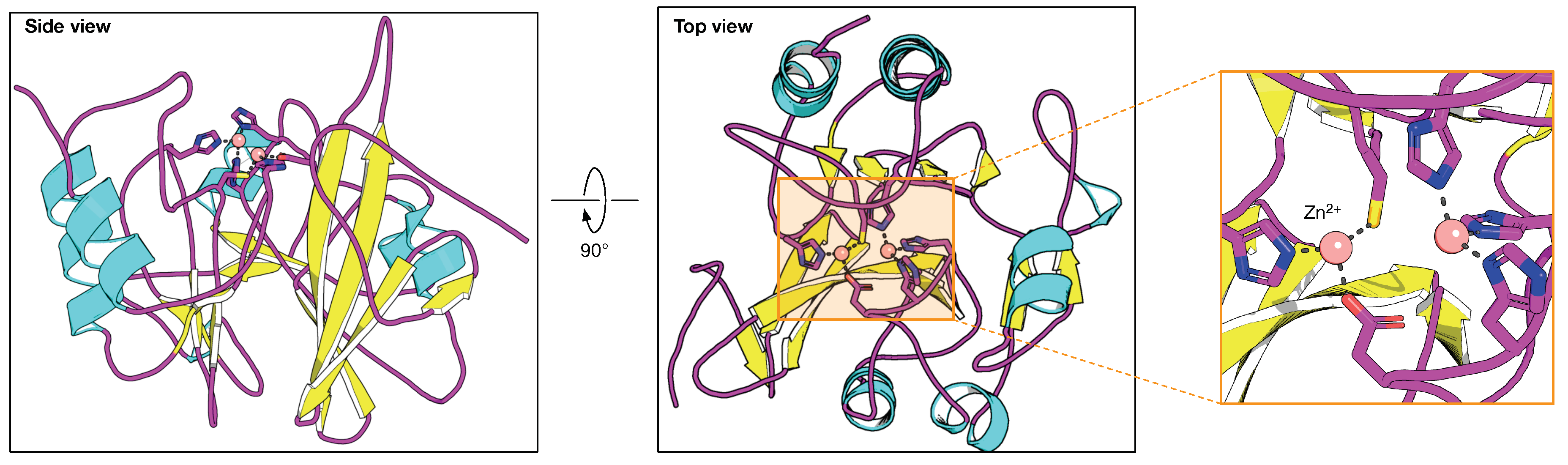
2.2. Class B -Lactamase
2.3. Class C -Lactamase
2.4. Class D -Lactamase
3. -Lactamase Inhibitors
3.1. Serine -Lactamase (Class A, C and D) Inhibitors
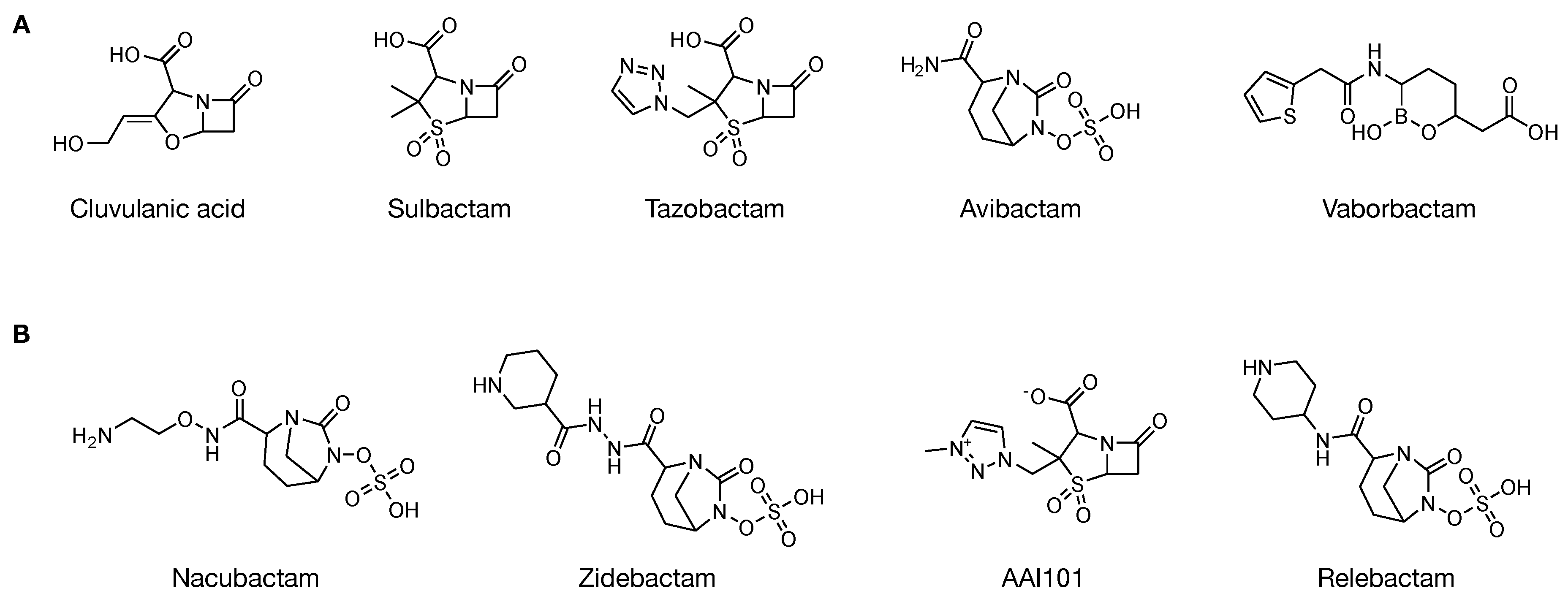
3.1.1. -Lactam/-Lactamase Inhibitors
3.1.2. Non--Lactam/-Lactamase Inhibitors
3.2. Class B -Lactamase Inhibitors
3.3. Resistance to -Lactamase Inhibitors
4. -Lactamase Inhibitor Proteins
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Antibiotic Resistance. 2018. Available online: http://www.who.int/en/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 15 June 2018).
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Review on Antimicrobial Resistance; Wellcome Trust: London, UK, 2016. [Google Scholar]
- O’Neill, J. Securing New Drugs for Future Generations: The Pipeline of Antibiotics; Review on Antimicrobial Resistance; Wellcome Trust: London, UK, 2015. [Google Scholar]
- World Health Organization. Antimicrobial Resistance. 2018. Available online: http://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 15 June 2018).
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Fatima, J.; Shakil, S.; Rizvi, S.M.D.; Kamal, M.A. Antibiotic resistance and extended spectrum β-lactamases: Types, epidemiology and treatment. Saudi J. Biol. Sci. 2015, 22, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Ferri, M.; Ranucci, E.; Romagnoli, P.; Giaccone, V. Antimicrobial resistance: A global emerging threat to public health systems. Crit. Rev. Food Sci. Nutr. 2017, 57, 2857–2876. [Google Scholar] [CrossRef] [PubMed]
- Wilke, M.S.; Lovering, A.L.; Strynadka, N.C.J. β-lactam antibiotic resistance: A current structural perspective. Curr. Opin. Microbiol. 2005, 8, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.; van Duin, D. Novel beta-lactamase inhibitors: Unlocking their potential in therapy. Drugs 2017, 77, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Kahne, D.; Kishony, R. Distinct single-cell morphological dynamics under β-lactam antibiotics. Mol. Cell 2012, 48, 705–712. [Google Scholar] [CrossRef] [PubMed]
- King, D.T.; Sobhanifar, S.; Strynadka, N.C.J. One ring to rule them all: Current trends in combating bacterial resistance to the β-lactams. Protein Sci. 2016, 25, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Frère, J.M.; Joris, B.; Granier, B.; Matagne, A.; Jacob, F.; Bourguignon-Bellefroid, C. Diversity of the mechanisms of resistance to β-lactam antibiotics. Res. Microbiol. 1991, 142, 705–710. [Google Scholar] [CrossRef]
- Pratt, R.F.; McLeish, M.J. Structural relationship between the active sites of β-lactam-recognizing and amidase signature enzymes: Convergent evolution? Biochemistry 2010, 49, 9688–9697. [Google Scholar] [CrossRef] [PubMed]
- Rudgers, G.W.; Palzkill, T. Protein minimization by random fragmentation and selection. Protein Eng. 2001, 14, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.S.; Apisarnthanarak, A.; Hsu, L.Y. Mechanisms of β-lactam antimicrobial resistance and epidemiology of major community- and healthcare-associated multidrug-resistant bacteria. Adv. Drug Deliv. Rev. 2014, 78, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Sandanayaka, V.P.; Prashad, A.S. Resistance to β-lactam antibiotics: structure and mechanism based design of β-lactamase inhibitors. Curr. Med. Chem. 2002, 9, 1145–1165. [Google Scholar] [CrossRef] [PubMed]
- Ambler, R.P. The structure of β-lactamases. Philos. Trans. R. Soc. B 1980, 289, 321–331. [Google Scholar] [CrossRef]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob. Agents Chemother. 1995, 39, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, R.A. β-Lactamases: A focus on current challenges. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Pitout, J.D.D.; Laupland, K.B. Extended-spectrum β-lactamase-producing Enterobacteriaceae: An emerging public-health concern. Lancet Infect. Dis. 2008, 8, 159–166. [Google Scholar] [CrossRef]
- Paterson, D.L.; Bonomo, R.A. Extended-spectrum β-lactamases: A clinical update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef] [PubMed]
- Rawat, D.; Nair, D. Extended-spectrum β-lactamases in Gram negative bacteria. J. Glob. Infect. Dis. 2010, 2, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.A. Extended-spectrum β-lactamases in the 21st century: Characterization, epidemiology, and detection of this important resistance threat. Clin. Microbiol. Rev. 2001, 14, 933–951. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Jacoby, G.A. Updated functional classification of β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Ali, T.; Ali, I.; Khan, N.A.; Han, B.; Gao, J. The growing genetic and functional diversity of extended spectrum β-lactamases. Biomed. Res. Int. 2018, 2018, 9519718. [Google Scholar] [CrossRef] [PubMed]
- Hanes, M.S.; Jude, K.M.; Berger, J.M.; Bonomo, R.A.; Handel, T.M. Structural and biochemical characterization of the interaction between KPC-2 β-lactamase and β-lactamase inhibitor protein. Biochemistry 2009, 48, 9185–9193. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Naas, T.; Poirel, L. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.H.; Hu, Z.Q. Epidemiology and genetics of CTX-M extended-spectrum β-lactamases in Gram-negative bacteria. Crit. Rev. Microbiol. 2013, 39, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Tzouvelekis, L.S.; Tzelepi, E.; Tassios, P.T.; Legakis, N.J. CTX-M-type β-lactamases: An emerging group of extended-spectrum enzymes. Int. J. Antimicrob. Agents 2000, 14, 137–142. [Google Scholar] [CrossRef]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile β-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Price, L.S.; Poirel, L.; Bonomo, R.A.; Schwaber, M.J.; Daikos, G.L.; Cormican, M.; Cornaglia, G.; Garau, J.; Gniadkowski, M.; Hayden, M.K.; et al. Clinical epidemiology of the global expansion of Klebsiella pneumoniae carbapenemases. Lancet Infect. Dis. 2013, 13, 785–796. [Google Scholar] [CrossRef]
- Nordmann, P.; Poirel, L. The difficult-to-control spread of carbapenemase producers among Enterobacteriaceae worldwide. Clin. Microbiol. Infect. 2014, 20, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Garau, G.; García-Sáez, I.; Bebrone, C.; Anne, C.; Mercuri, P.; Galleni, M.; Frère, J.M.; Dideberg, O. Update of the standard numbering scheme for class B β-lactamases. Antimicrob. Agents Chemother. 2004, 48, 2347–2349. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.F.; Mobashery, S. Three decades of the class A β-lactamase acyl-enzyme. Curr. Protein Pept. Sci. 2009, 10, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Palzkill, T. Metallo-β-lactamase structure and function. Ann. N. Y. Acad. Sci. 2013, 1277, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.R.; Toleman, M.A.; Poirel, L.; Nordmann, P. Metallo-β-lactamases: The quiet before the storm? Clin. Microbiol. Rev. 2005, 18, 306–325. [Google Scholar] [CrossRef] [PubMed]
- Zmarlicka, M.T.; Nailor, M.D.; Nicolau, D.P. Impact of the New Delhi metallo-beta-lactamase on beta-lactam antibiotics. Infect. Drug Resist. 2015, 8, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Yong, D.; Toleman, M.A.; Giske, C.G.; Cho, H.S.; Sundman, K.; Lee, K.; Walsh, T.R. Characterization of a new metallo-β-lactamase gene, blaNDM-1, and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob. Agents Chemother. 2009, 53, 5046–5054. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.U.; Maryam, L.; Zarrilli, R. Structure, genetics and worldwide spread of New Delhi metallo-β-lactamase (NDM): A threat to public health. BMC Microbiol. 2017, 17, 101. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Valladares, M.; Felici, A.; Weber, G.; Adolph, H.W.; Zeppezauer, M.; Rossolini, G.M.; Amicosante, G.; Frère, J.M.; Galleni, M. Zn(II) dependence of the Aeromonas hydrophila AE036 metallo-β-lactamase activity and stability. Biochemistry 1997, 36, 11534–11541. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fast, W.; Valentine, A.M.; Benkovic, S.J. Metallo-β-lactamase: Structure and mechanism. Curr. Opin. Chem. Biol. 1999, 3, 614–622. [Google Scholar] [CrossRef]
- Philippon, A.; Arlet, G.; Jacoby, G.A. Plasmid-determined AmpC-type β-lactamases. Antimicrob. Agents Chemother. 2002, 46, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A. AmpC β-lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Mohamudha Parveen, R.; Harish, B.N.; Parija, S.C. AmpC β-lactamases among Gram negative clinical isolates from a tertiary hospital, South India. Brazil. J. Microbiol. 2010, 41, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Thomson, K.S. Extended-spectrum-β-lactamase, AmpC, and carbapenemase issues. J. Clin. Microbiol. 2010, 48, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Jaurin, B.; Grundström, T. ampC cephalosporinase of Escherichia coli K-12 has a different evolutionary origin from that of β-lactamases of the penicillinase type. Proc. Nat. Acad. Sci. USA 1981, 78, 4897–4901. [Google Scholar] [CrossRef] [PubMed]
- Knott-Hunziker, V.; Petursson, S.; Jayatilake, G.S.; Waley, S.G.; Jaurin, B.; Grundström, T. Active sites of β-lactamases. The chromosomal β-lactamases of Pseudomonas aeruginosa and Escherichia coli. Biochem. J. 1982, 201, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Papp-Wallace, K.M.; Bonomo, R.A. New β-lactamase inhibitors: A therapeutic renaissance in an MDR world. Antimicrob. Agents Chemother. 2014, 58, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Minasov, G.; Roth, T.A.; Prati, F.; Shoichet, B.K. The deacylation mechanism of AmpC β-lactamase at ultrahigh resolution. J. Am. Chem. Soc. 2006, 128, 2970–2976. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Naas, T.; Nordmann, P. Diversity, epidemiology, and genetics of class D β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Antunes, N.T.; Fisher, J.F. Acquired class D β-lactamases. Antibiotics 2014, 3, 398–434. [Google Scholar] [CrossRef] [PubMed]
- Antunes, N.T.; Lamoureaux, T.L.; Toth, M.; Stewart, N.K.; Frase, H.; Vakulenko, S.B. Class D β-lactamases: Are they all carbapenemases? Antimicrob. Agents Chemother. 2014, 58, 2119–2125. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.A.; Amyes, S.G.B. OXA β-lactamases. Clin. Microbiol. Rev. 2014, 27, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Antunes, N.T.; Stewart, N.K.; Toth, M.; Kumarasiri, M.; Chang, M.; Mobashery, S.; Vakulenko, S.B. Structural basis for carbapenemase activity of the OXA-23 β-lactamase from Acinetobacter baumannii. Chem. Biol. 2013, 20. [Google Scholar] [CrossRef] [PubMed]
- Reading, C.; Cole, M. Clavulanic acid: A beta-lactamase-inhiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef] [PubMed]
- English, A.R.; Retsema, J.A.; Girard, A.E.; Lynch, J.E.; Barth, W.E. CP-45,899, a beta-lactamase inhibitor that extends the antibacterial spectrum of beta-lactams: Initial bacteriological characterization. Antimicrob. Agents Chemother. 1978, 14, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Belasco, J.G.; Charnas, R.L.; Khosla, S.; Knowles, J.R. β-lactamase inactivation by mechanism-based reagents. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1980, 289, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Geddes, A.M.; Klugman, K.P.; Rolinson, G.N. Introduction: Historical perspective and development of amoxicillin/clavulanate. Int. J. Antimicrob. Agents 2007, 30 (Suppl. 2), S109–S112. [Google Scholar] [CrossRef] [PubMed]
- Buynak, J.D. Understanding the longevity of the β-lactam antibiotics and of antibiotic/β-lactamase inhibitor combinations. Biochem. Pharmacol. 2006, 71, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Monnaie, D.; Frere, J.M. Interaction of clavulanate with class C β-lactamases. FEBS Lett. 1993, 334, 269–271. [Google Scholar] [CrossRef]
- Page, M.G.P. β-lactamase inhibitors. Drug Resist. Updat. 2000, 3, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.J.; Cramp, R.; Winstanley, D.J.; Knowles, D.J. Comparative activities of clavulanic acid, sulbactam, and tazobactam against clinically important β-lactamases. Antimicrob. Agents Chemother. 1994, 38, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, A.; Dupuis-Hamelin, C.; Steier, V.; Delachaume, C.; Seys, C.; Stachyra, T.; Fairley, M.; Guitton, M.; Lampilas, M. In vitro activity of AVE1330A, an innovative broad-spectrum non-β-lactam β-lactamase inhibitor. J. Antimicrob. Chemother. 2004, 54, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Abboud, M.I.; Markoulides, M.S.; Brem, J.; Schofield, C.J. The road to avibactam: The first clinically useful non-β-lactam working somewhat like a β-lactam. Future Med. Chem. 2016, 8, 1063–1084. [Google Scholar] [CrossRef] [PubMed]
- Temkin, E.; Torre-Cisneros, J.; Beovic, B.; Benito, N.; Giannella, M.; Gilarranz, R.; Jeremiah, C.; Loeches, B.; Machuca, I.; Jiménez-Martín, M.J. Ceftazidime-Avibactam as salvage therapy for infections caused by carbapenem-resistant organisms. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Lahiri, S.D.; Johnstone, M.R.; Ross, P.L.; McLaughlin, R.E.; Olivier, N.B.; Alm, R.A. Avibactam and class C β-lactamases: Mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob. Agents Chemother. 2014, 58, 5704–5713. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.D.; Mangani, S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updat. 2018, 36, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Werner, J.P.; Mitchell, J.M.; Taracila, M.A.; Bonomo, R.A.; Powers, R.A. Exploring the potential of boronic acids as inhibitors of OXA-24/40 β-lactamase. Protein Sci. 2017, 26, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Powers, R.A.; Blasczcak, L.C.; Wu, C.Y.E.; Prati, F.; Shoichet, B.K. Energetic, structural, and antimicrobial analyses of β-lactam side chain recognition by β-lactamases. Chem. Biol. 2001, 8, 17–31. [Google Scholar] [CrossRef]
- Patera, A.; Blaszczak, L.C.; Shoichet, B.K. Crystal structures of substrate and inhibitor complexes with AmpC β-lactamase: Possible implications for substrate-assisted catalysis. J. Am. Chem. Soc. 2000, 122, 10504–10512. [Google Scholar] [CrossRef]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jiménez-Castellanos, J.C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.G.; Schofield, C.J. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun. 2016, 7, 12406. [Google Scholar] [CrossRef] [PubMed]
- Cahill, S.T.; Cain, R.; Wang, D.Y.; Lohans, C.T.; Wareham, D.W.; Oswin, H.P.; Mohammed, J.; Spencer, J.; Fishwick, C.W.G.; McDonough, M.A.; Schofielda, C.J.; Brem, J. Cyclic boronates inhibit all classes of β-lactamases. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Bou, G.; Santillana, E.; Sheri, A.; Beceiro, A.; Sampson, J.M.; Kalp, M.; Bethel, C.R.; Distler, A.M.; Drawz, S.M.; Pagadala, S.R.R.; et al. Design, synthesis, and crystal structures of 6-alkylidene-2’-substituted penicillanic acid sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J. Am. Chem. Soc. 2010, 132, 13320–13331. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bethel, C.R.; Doppalapudi, V.R.; Sheri, A.; Pagadala, S.R.R.; Hujer, A.M.; Skalweit, M.J.; Anderson, V.E.; Chen, S.G.; Buynak, J.D.; et al. Penicillin sulfone inhibitors of class D β-lactamases. Antimicrob. Agents Chemother. 2010, 54, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Danishuddin, M.; Khan, A.; Faheem, M.; Kalaiarasan, P.; Hassan Baig, M.; Subbarao, N.; Khan, A.U. Structure-based screening of inhibitors against KPC-2: Designing potential drug candidates against multidrug-resistant bacteria. J. Biomol. Struct. Dyn. 2014, 32, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, C.M.; Wright, G.D. Inhibitors of metallo-β-lactamases. Curr. Opin. Microbiol. 2017, 39, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, K.H.M.E.; Martin, N.I. Thiol-containing metallo-β-lactamase inhibitors resensitize resistant Gram-negative bacteria to meropenem. ACS Infect. Dis. 2017, 3, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Li, G.B.; Abboud, M.I.; Brem, J.; Someya, H.; Lohans, C.T.; Yang, S.Y.; Spencer, J.; Wareham, D.W.; McDonough, M.A.; Schofield, C.J. NMR-filtered virtual screening leads to non-metal chelating metallo-β-lactamase inhibitors. Chem. Sci. 2017, 8, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Hinchliffe, P.; Tanner, C.A.; Krismanich, A.P.; Labbé, G.; Goodfellow, V.J.; Marrone, L.; Desoky, A.Y.; Calvopiña, K.; Whittle, E.E.; Zeng, F.; et al. Structural and kinetic studies of the potent inhibition of metallo-β-lactamases by 6-phosphonomethylpyridine-2-carboxylates. Biochemistry 2018, 57, 1880–1892. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Game changers: New β-lactamase inhibitor combinations targeting antibiotic resistance in Gram-negative bacteria. ACS Infect. Dis. 2018, 4, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Doran, J.L.; Leskiw, B.K.; Aippersbach, S.; Jensen, S.E. Isolation and characterization of a β-lactamase-inhibitory protein from Streptomyces clavuligerus and cloning and analysis of the corresponding gene. J. Bacteriol. 1990, 172, 4909–4918. [Google Scholar] [CrossRef] [PubMed]
- Strynadka, N.C.; Jensen, S.E.; Johns, K.; Blanchard, H.; Page, M.; Matagne, A.; Frère, J.M.; James, M.N. Structural and kinetic characterization of a β-lactamase-inhibitor protein. Nature 1994, 368, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Chow, D.C.; Rice, K.; Huang, W.; Atmar, R.L.; Palzkill, T. Engineering specificity from broad to narrow: Design of a β-lactamase inhibitory protein (BLIP) variant that exclusively binds and detects KPC β-lactamase. ACS Infect. Dis. 2016, 2, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Palzkill, T. Determinants of binding affinity and specificity for the interaction of TEM-1 and SME-1 β-lactamase with β-lactamase inhibitory protein. J. Biol. Chem. 2003, 278, 45706–45712. [Google Scholar] [CrossRef] [PubMed]
- Strynadka, N.C.J.; Eisenstein, M.; Katchalski-Katzir, E.; Shoichet, B.K.; Kuntz, I.D.; Abagyan, R.; Totrov, M.; Janin, J.; Cherfils, J.; Zimmerman, F. Molecular docking programs successfully predict the binding of a β-lactamase inhibitory protein to TEM-1 β-lactamase. Nat. Struct. Mol. Biol. 1996, 3, 233–239. [Google Scholar] [CrossRef]
- Petrosino, J.; Rudgers, G.; Gilbert, H.; Palzkill, T. Contributions of aspartate 49 and phenylalanine 142 residues of a tight binding inhibitory protein of β-lactamases. J. Biol. Chem. 1999, 274, 2394–2400. [Google Scholar] [CrossRef] [PubMed]
- Selzer, T.; Albeck, S.; Schreiber, G. Rational design of faster associating and tighter binding protein complexes. Nat. Struct. Biol. 2000, 7, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Adamski, C.J.; Palzkill, T. Systematic substitutions at BLIP position 50 result in changes in binding specificity for class A β-lactamases. BMC Biochem. 2017, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.G.; Park, H.U.; Lee, H.S.; Kim, H.T.; Lee, K.J. New β-lactamase inhibitory protein (BLIP-I) from Streptomyces exfoliatus SMF19 and its roles on the morphological differentiation. J. Biol. Chem. 2000, 275, 16851–16856. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.; Park, H.U.; De Castro, L.; Kang, S.G.; Lee, H.S.; Jensen, S.; Lee, K.J.; Strynadka, N.C. Crystal structure and kinetic analysis of β-lactamase inhibitor protein-II in complex with TEM-1 β-lactamase. Nat. Struct. Biol. 2001, 8, 848–852. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Llarena, F.J.; Liras, P.; Rodríguez-García, A.; Martín, J.F. A regulatory gene (ccaR) required for cephamycin and clavulanic acid production in Streptomyces clavuligerus: Amplification results in overproduction of both β-lactam compounds. J. Bacteriol. 1997, 179, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Gretes, M.; Lim, D.C.; de Castro, L.; Jensen, S.E.; Kang, S.G.; Lee, K.J.; Strynadka, N.C.J. Insights into positive and negative requirements for protein-protein interactions by crystallographic analysis of the β-lactamase inhibitory proteins BLIP, BLIP-I, and BLP. J. Mol. Biol. 2009, 389, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Lee, K.J. Characteristics of β-lactamase-inhibiting proteins from Streptomyces exfoliatus SMF19. Appl. Environ. Microbiol. 1994, 60, 1029–1032. [Google Scholar] [PubMed]
- Huang, W.; Petrosino, J.; Palzkill, T. Display of functional β-lactamase inhibitory protein on the surface of M13 bacteriophage. Antimicrob. Agents Chemother. 1998, 42, 2893–2897. [Google Scholar] [PubMed]
- Albeck, S.; Schreiber, G. Biophysical Characterization of the Interaction of the β-Lactamase TEM-1 with its protein inhibitor BLIP. Biochem. 1999, 38, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhang, Z.; Palzkill, T. Design of potent β-lactamase inhibitors by phage display of β-lactamase inhibitory protein. J. Biol. Chem. 2000, 275, 14964–14968. [Google Scholar] [CrossRef] [PubMed]
- Rudgers, G.W.; Palzkill, T. Identification of residues in β-lactamase critical for binding β-lactamase inhibitory protein. J. Biol. Chem. 1999, 274, 6963–6971. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, W.A.; Locke, T.R.; Jensen, S.E. Resistance to β-lactamase inhibitor protein does not parallel resistance to clavulanic acid in TEM β-lactamase mutants. Antimicrob. Agents Chemother. 2002, 46, 3568–3573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Palzkill, T. Dissecting the protein-protein interface between β-lactamase inhibitory protein and class A β-lactamases. J. Biol. Chem. 2004, 279, 42860–42866. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.G.; Palzkill, T. Identification and characterization of β-lactamase inhibitor protein-II (BLIP-II) interactions with β-lactamases using phage display. Protein Eng. Des. Sel. 2010, 23, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Strynadka, N.C.; Jensen, S.E.; Alzari, P.M.; James, M.N. A potent new mode of β-lactamase inhibition revealed by the 1.7 Å X-ray crystallographic structure of the TEM-1-BLIP complex. Nat. Struct. Biol. 1996, 3, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Fryszczyn, B.G.; Adamski, C.J.; Brown, N.G.; Rice, K.; Huang, W.; Palzkill, T. Role of β-lactamase residues in a common interface for binding the structurally unrelated inhibitory proteins BLIP and BLIP-II. Protein Sci. 2014, 23, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.G.; Chow, D.C.; Palzkill, T. BLIP-II is a highly potent inhibitor of Klebsiella pneumoniae carbapenemase (KPC-2). Antimicrob. Agents Chemother. 2013, 57, 3398–3401. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Khait, R.; Schreiber, G. Low-stringency selection of TEM1 for BLIP shows interface plasticity and selection for faster binders. Proc. Natl. Acad. Sci. USA 2016, 113, 14982–14987. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Khait, R.; Schreiber, G. Selecting for fast protein-protein association as demonstrated on a random TEM1 yeast library binding BLIP. Biochemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Joughin, B.A.; Green, D.F.; Tidor, B. Action-at-a-distance interactions enhance protein binding affinity. Protein Sci. 2005, 14, 1363–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selzer, T.; Schreiber, G. New insights into the mechanism of protein-protein association. Proteins 2001, 45, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Meneksedag, D.; Dogan, A.; Kanlikilicer, P.; Ozkirimli, E. Communication between the active site and the allosteric site in class A β-lactamases. Computat. Biol. Chem. 2013, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Ozkirimli Olmez, E.; Budeyri, N.; Akbulut, B.S. Investigation of TEM-1 and SHV-1 β-lactamase ligand binding. In Proceedings of the 5th International Symposium on Health Informatics and Bioinformatics, Antalya, Turkey, 20–22 April 2010; pp. 167–172. [Google Scholar] [CrossRef]
- Fataftah, H.; Karain, W. Detecting protein atom correlations using correlation of probability of recurrence. Proteins 2014, 82, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Kozer, N.; Schreiber, G. Effect of crowding on protein-protein association rates: Fundamental differences between low and high mass crowding agents. J. Mol. Biol. 2004, 336, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.B.; Chui, K.S.; Chan, C.L.; Tsang, C.W.; Leung, Y.C. An efficient heat-inducible Bacillus subtilis bacteriophage 105 expression and secretion system for the production of the Streptomyces clavuligerus β-lactamase inhibitory protein (BLIP). J. Biotechnol. 2004, 108, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, K.A.; Thomson, J.M.; Corbett, K.D.; Bethel, C.R.; Berger, J.M.; Kirsch, J.F.; Bonomo, R.A.; Handel, T.M. Structural and computational characterization of the SHV-1 β-lactamase-β-lactamase inhibitor protein interface. J. Biol. Chem. 2006, 281, 26745–26753. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Cohen, M.; Schreiber, G. On the dynamic nature of the transition state for protein-protein association as determined by double-mutant cycle analysis and simulation. J. Mol. Biol. 2007, 371, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Z.; Palzkill, T.; Chow, D.C. Thermodynamic investigation of the role of contact residues of β-lactamase-inhibitory protein for binding to TEM-1 β-lactamase. J. Biol. Chem. 2007, 282, 17676–17684. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, K.A.; Hanes, M.S.; Thomson, J.M.; Antczak, A.J.; Berger, J.M.; Bonomo, R.A.; Kirsch, J.F.; Handel, T.M. Computational redesign of the SHV-1 β-lactamase/β-lactamase inhibitor protein interface. J. Mol. Biol. 2008, 382, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Palzkill, T.; Chow, D.C. Structural insight into the kinetics and DeltaCp of interactions between TEM-1 β-lactamase and β-lactamase inhibitory protein (BLIP). J. Biol. Chem. 2009, 284, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Huang, W.; Chow, D.C.; Palzkill, T. Fine mapping of the sequence requirements for binding of β-lactamase inhibitory protein (BLIP) to TEM-1 β-lactamase using a genetic screen for BLIP function. J. Mol. Biol. 2009, 389, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Hanes, M.S.; Ratcliff, K.; Marqusee, S.; Handel, T.M. Protein-protein binding affinities by pulse proteolysis: Application to TEM-1/BLIP protein complexes. Protein Sci. 2010, 19, 1996–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, N.G.; Chow, D.C.; Sankaran, B.; Zwart, P.; Prasad, B.V.V.; Palzkill, T. Analysis of the binding forces driving the tight interactions between β-Lactamase inhibitory protein-II (BLIP-II) and class A β-Lactamases. J. Biol. Chem. 2011, 286, 32723–32735. [Google Scholar] [CrossRef] [PubMed]
- Fryszczyn, B.G.; Brown, N.G.; Huang, W.; Balderas, M.A.; Palzkill, T. Use of periplasmic target protein capture for phage display engineering of tight-binding protein-protein interactions. Protein Eng. Des. Sel. 2011, 24, 819–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanes, M.S.; Reynolds, K.A.; McNamara, C.; Ghosh, P.; Bonomo, R.A.; Kirsch, J.F.; Handel, T.M. Specificity and cooperativity at β-lactamase position 104 in TEM-1/BLIP and SHV-1/BLIP interactions. Proteins 2011, 79, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chow, D.C.; Huang, W.; Palzkill, T. Identification of a β-lactamase inhibitory protein variant that is a potent inhibitor of Staphylococcus PC1 β-lactamase. J. Mol. Biol. 2011, 406, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Phillip, Y.; Kiss, V.; Schreiber, G. Protein-binding dynamics imaged in a living cell. Proc. Nat. Acad. Sci. USA 2012, 109, 1461–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillip, Y.; Harel, M.; Khait, R.; Qin, S.; Zhou, H.X.; Schreiber, G. Contrasting factors on the kinetic path to protein complex formation diminish the effects of crowding agents. Biophys. J. 2012, 103, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Alaybeyoglu, B.; Akbulut, B.S.; Ozkirimli, E. A novel chimeric peptide with antimicrobial activity. J. Pept. Sci. 2015, 21, 294–301. [Google Scholar] [CrossRef] [PubMed]
- De Kraker, M.E.A.; Stewardson, A.J.; Harbarth, S. Will 10 million people die a year due to antimicrobial resistance by 2050? PLoS Med. 2016, 13, e1002184. [Google Scholar]
- Gomes, B.; Augusto, M.T.; Felício, M.R.; Hollmann, A.; Franco, O.L.; Gonçalves, S.; Santos, N.C. Designing improved active peptides for therapeutic approaches against infectious diseases. Biotechnol. Adv. 2018, 36, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Sable, R.; Parajuli, P.; Jois, S. Peptides, peptidomimetics, and polypeptides from marine sources: A wealth of natural sources for pharmaceutical applications. Mar. Drugs 2017, 15, 124. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, P.; Dortet, L.; Poirel, L. Carbapenem resistance in Enterobacteriaceae: Here is the storm! Trends Mol. Med. 2012, 18, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Melo, M.N.; Castanho, M.A.R.B. Cell-penetrating peptides and antimicrobial peptides: How different are they? Biochem. J. 2006, 399, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nantasenamat, C.; Prachayasittikul, V. Maximizing computational tools for successful drug discovery. Expert Opin. Drug Discov. 2015, 10, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]





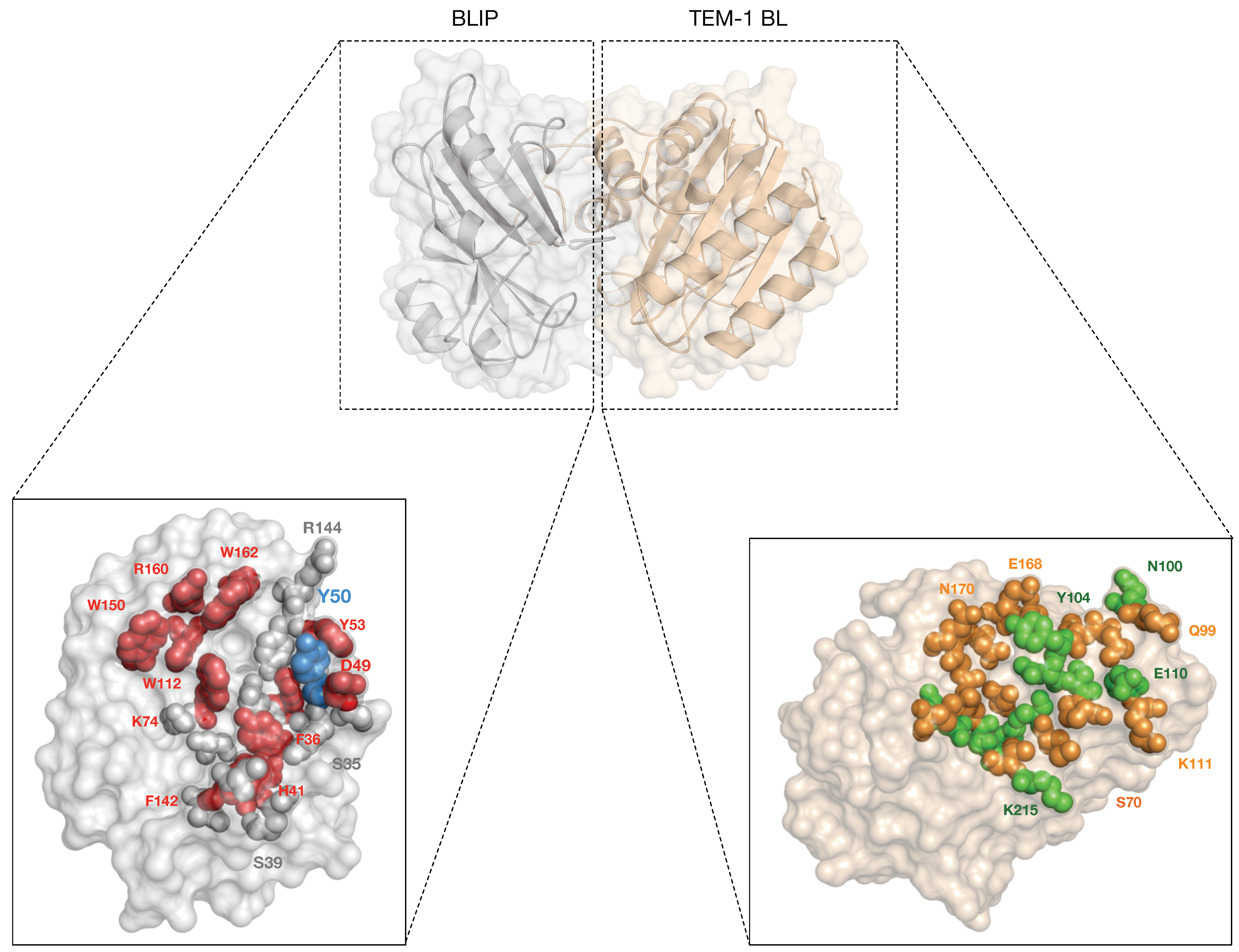

{kind=link}
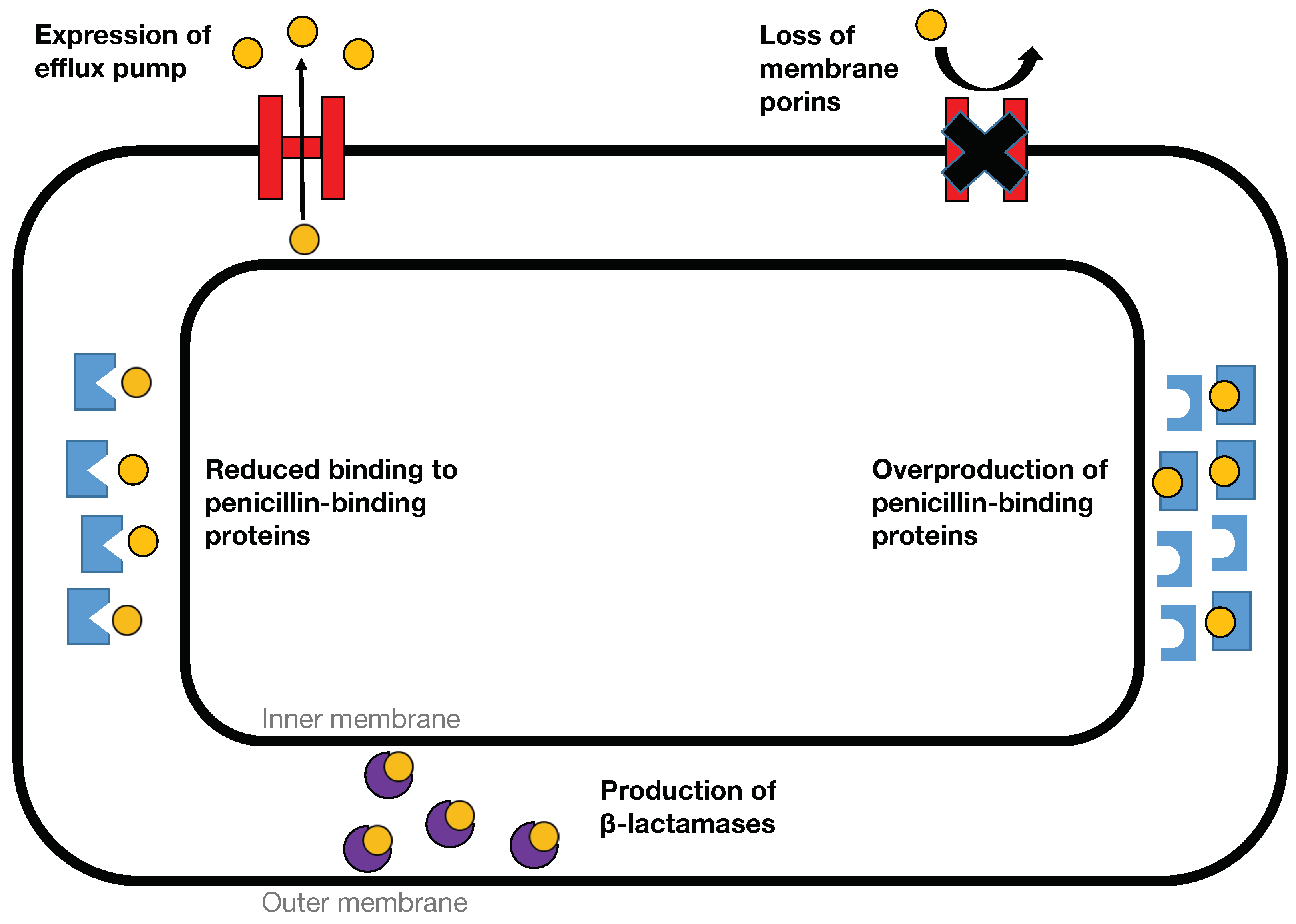{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
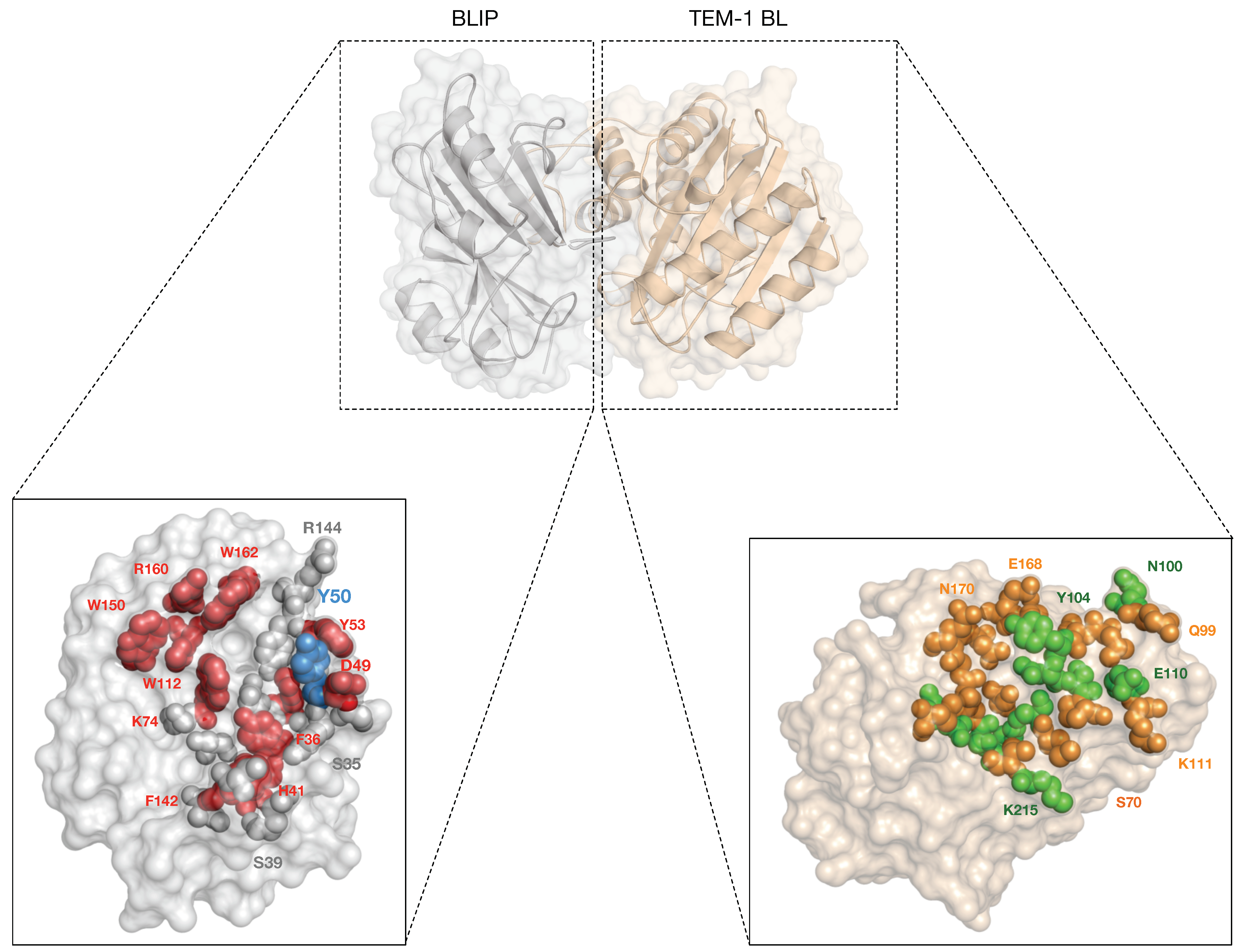{kind=link}
| Year | BLIP Type | Protein Source | Binding Affinity | Detection Method | BL Target | Ref. |
|---|---|---|---|---|---|---|
| 1994 | BLIP | Secreted protein from S. claviligerus | = 18 pM − 12 M | Enzyme inhibition | Class A-D and PBPs | [86] |
| 1994 | BLIP-I, BLIP-II | Secreted protein from S. exfoliatus SMF19 | BLIP-I: = 0.062 nM, BLIP-II: K = 0.274 M | Enzyme inhibition | Bacto Penase | [98] |
| 1996 | BLIP | Secreted protein from S. claviligerus | TEM-1: = 0.6 nM | Enzyme inhibition | TEM-1 | [106] |
| 1998 | BLIP | Phage display system | IC = 1 nM | Phage ELISA | TEM-1 | [99] |
| 1999 | BLIP | E. coli expression system | Enzyme inhibition: = 0.4 nM, FQT: = 0.3 nM, SPR: = 15 nM | Enzyme inhibition, FQT, SPR | TEM-1 | [100] |
| 1999 | BLIP | E. coli expression system | TEM-1: = 0.11 nM, SHV-1: K = 1 M | Enzyme inhibition | TEM-1, SHV-1 | [90] |
| 1999 | BLIP | E. coli expression system | = 0.22 nM | Enzyme inhibition | TEM-1 | [102] |
| 2000 | BLIP | E. coli expression system | = 0.12 nM | Enzyme inhibition | TEM-1 | [101] |
| 2000 | BLIP-I | Secreted protein from S. exfoliatus SMF19 and E. coli expression system | Secreted protein: = 0.047 nM, Recombinant BLIP-I: K = 0.062 nM | Enzyme inhibition | TEM-1 | [94] |
| 2000 | BLIP | E. coli expression system | K = 1.25 nM | Enzyme inhibition by SFF, SPR | TEM-1 | [91] |
| 2001 | BLIP-II | Secreted protein from S. exfoliatus SMF19 | = 0.0272 nM | Enzyme inhibition | TEM-1 | [95] |
| 2001 | BLIP | Phage display system | BLIP peptide (residue C30-D49): = 352 M | Enzyme inhibition | TEM-1 | [15] |
| 2002 | BLIP | Secreted protein from S. claviligerus | = 2.76 nM | Enzyme inhibition | TEM-1 | [103] |
| 2003 | BLIP | E. coli expression system | TEM-1: = 0.5 nM, SME-1: = 2.4 nM | Enzyme inhibition | TEM-1 and SME-1 | [88] |
| 2004 | BLIP | E. coli expression system | = 4.4 ×10 M·s | Enzyme inhibition by SFF | TEM-1 | [116] |
| 2004 | BLIP | B. subtilis expression system | = 0.31 nM | Enzyme inhibition | TEM-1 | [117] |
| 2004 | BLIP | E. coli expression system | TEM-1: = 0.5 nM, SME-1: = 2.4 nM, SHV-1: = 1 M, Bla1: = 2.5 nM | Enzyme inhibition | TEM-1, SME-1, SHV-1 and Bla1 | [104] |
| 2006 | BLIP | E. coli expression system | TEM-1: = 0.32 nM, SHV-1: = 1.25 M | Enzyme inhibition | TEM-1, SHV-1 | [118] |
| 2007 | BLIP | E. coli expression system | = 3.3 ×10 M·s | Enzyme inhibition by SFF | TEM-1 | [119] |
| 2007 | BLIP | E. coli expression system | TEM-1: = 0.5 nM | Enzyme inhibition | TEM-1 | [120] |
| 2008 | BLIP | E. coli expression system | TEM-1: = 1.3 nM, SHV-1: = 1.72 M | Enzyme inhibition | TEM-1, SHV-1 | [121] |
| 2009 | BLIP, BLIP-I, BLIP-like protein | E. coli expression system | BLIP: = 0.1–0.6 nM, BLIP-I: = 0.05 nM | Enzyme inhibition | TEM-1 | [97] |
| 2009 | BLIP | E. coli expression system | KPC-2: = 84 pM, KPC-3: = 250 pM | Enzyme inhibition | KPC-2, KPC-3 | [28] |
| 2009 | BLIP | E. coli expression system | = 2×10 M·s | Enzyme inhibition by SFF | TEM-1 | [122] |
| 2009 | BLIP | E. coli expression system | TEM-1: = 0.5 nM | Enzyme inhibition | TEM-1 | [123] |
| 2010 | BLIP-II | Phage display system and E. coli expression system | TEM-1: IC = 320 pM, TEM-1: = 2.5 pM, SME-1: = 8.4 pM, SHV-1: = 12 pM, Bla1: ≤ 25 pM, CTX-M14: = 9.1 pM, PC1: ≤ 16 pM | Phage ELISA, Enzyme inhibition | TEM-1, SHV-1, Bla 1, SME-1, CTX-M14, PC1 | [105] |
| 2010 | BLIP | E. coli expression system | = 1.7 nM | Enzyme inhibition | TEM-1 | [124] |
| 2011 | BLIP-II | E. coli expression system | TEM-1: = 0.79 pM, PC1: = 7.8 pM, SHV-1: = 30 pM, Bla1: = 1.1 pM | Enzyme inhibition by SFF | TEM-1, PC1, SHV-1, Bla1 | [125] |
| 2011 | BLIP | Phage display system and E. coli expression system | = 0.5 nM, = 0.4 nM | Phage ELISA, Enzyme inhibition | TEM-1 | [126] |
| 2011 | BLIP | E. coli expression system | TEM-1: = 3.2 nM, SHV-1: = 1.8 M | Enzyme inhibition | TEM-1, SHV-1 | [127] |
| 2011 | BLIP | E. coli expression system | = 350 nM, = 380 nM, = 1×10 M·s | Enzyme inhibition by SFF, ITC | PC1 | [128] |
| 2012 | BLIP | E. coli expression system | = 3×10 M·s | Enzyme inhibition by SFF | TEM-1 | [129] |
| 2012 | BLIP | E. coli expression system | = 2.9×10 M·s | Enzyme inhibition by SFF | TEM-1 | [130] |
| 2013 | BLIP-II | E. coli expression system | = 76 fM | Enzyme inhibition | KPC-2 | [108] |
| 2014 | BLIP, BLIP-II | E. coli expression system | BLIP: = 230 pM, BLIP-II: = 1.5 pM | Enzyme inhibition by SFF | TEM-1 | [107] |
| 2015 | BLIP | Peptide synthesis | BLIP peptide (residue H45-Y53): = 58 M | Enzyme inhibition | TEM-1 | [131] |
| 2016 | BLIP | E. coli expression system | TEM-1: = 0.5 nM, KPC-2: = 1.2 nM, SHV-1: = 1.1 M, CTX-M14: = 810 nM, SME-1: = 2.4 nM | Enzyme inhibition | TEM-1, KPC-2, SHV-1, CTX-M14, SME-1 | [87] |
| 2016 | BLIP | E. coli expression system | = 28 nM | Enzyme inhibition by SFF, SPR | TEM-1 | [109] |
| 2017 | BLIP | E. coli expression system | TEM-1: = 0.5 nM, SHV-1: = 1.13 M, KPC-2: = 1.5 nM, Bla1: = 2.5 nM | Enzyme inhibition | TEM-1, SHV-1, KPC-2, Bla1 | [93] |
| 2018 | BLIP | E. coli expression system | = 0.57 nM | Enzyme inhibition by SFF, SPR | TEM-1 | [110] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eiamphungporn, W.; Schaduangrat, N.; Malik, A.A.; Nantasenamat, C. Tackling the Antibiotic Resistance Caused by Class A β-Lactamases through the Use of β-Lactamase Inhibitory Protein. Int. J. Mol. Sci. 2018, 19, 2222. https://doi.org/10.3390/ijms19082222
Eiamphungporn W, Schaduangrat N, Malik AA, Nantasenamat C. Tackling the Antibiotic Resistance Caused by Class A β-Lactamases through the Use of β-Lactamase Inhibitory Protein. International Journal of Molecular Sciences. 2018; 19(8):2222. https://doi.org/10.3390/ijms19082222
Chicago/Turabian StyleEiamphungporn, Warawan, Nalini Schaduangrat, Aijaz Ahmad Malik, and Chanin Nantasenamat. 2018. "Tackling the Antibiotic Resistance Caused by Class A β-Lactamases through the Use of β-Lactamase Inhibitory Protein" International Journal of Molecular Sciences 19, no. 8: 2222. https://doi.org/10.3390/ijms19082222
APA StyleEiamphungporn, W., Schaduangrat, N., Malik, A. A., & Nantasenamat, C. (2018). Tackling the Antibiotic Resistance Caused by Class A β-Lactamases through the Use of β-Lactamase Inhibitory Protein. International Journal of Molecular Sciences, 19(8), 2222. https://doi.org/10.3390/ijms19082222