mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases


Abstract
1. Introduction
1.1. Senescence
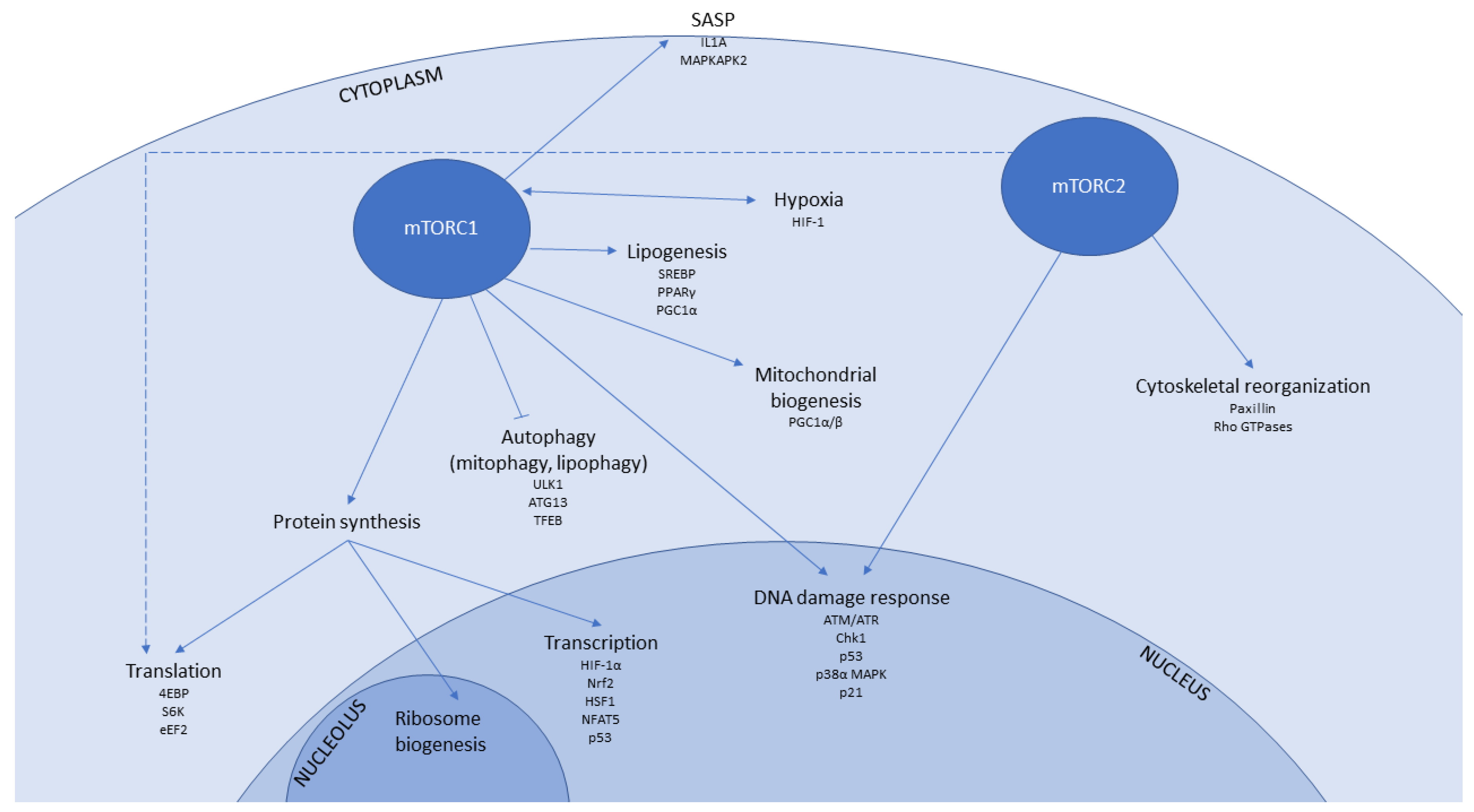
1.2. mTOR Signalling in Senescence and Ageing
1.3. mTOR-Associated Pathways That Contribute to Senescence and Ageing
1.3.1. Transcription
1.3.2. Protein Translation
1.3.3. Autophagy
1.3.4. Mitochondrial Function and Biogenesis
1.3.5. Hypoxia
1.3.6. Immunomodulatory Signalling
1.3.7. DNA Damage Response
1.3.8. Lipid Metabolism
1.4. Rapamycin and Other mTOR Inhibitors
2. Ageing and Age-Related Pathologies Amenable to Treatment by mTOR Inhibition
2.1. Ageing
2.2. Immunosenescence
2.3. Age-Related Neurodegeneration
2.3.1. Alzheimer’s Disease
2.3.2. Huntington’s Disease
2.3.3. Parkinson’s Disease
2.4. Age-Related Blindness: AMD
2.5. Musculoskeletal Disorders
2.5.1. Sarcopenia and Muscle Wasting
2.5.2. Osteoporosis
2.5.3. Rheumatoid Arthritis
2.5.4. Diabetic Bone Fragility
2.6. Cardiovascular Disease
2.7. Kidney Disease
2.7.1. Adult Polycystic Kidney Disease
2.7.2. Diabetic Nephropathy
2.8. Age-Related Cancer
3. Perspectives
3.1. Balancing Efficacy Against Side Effects
3.2. Monitoring Therapeutic Outcomes: The Need for Ageing Biomarkers
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| 4EBP1 | eIF4E binding protein |
| 53BP1 | p53 binding protein 1 |
| Aβ | amyloid beta |
| AD | Alzheimer’s disease |
| ADPKD | adult polycystic kidney disease |
| Akt/PKB | protein kinase B |
| AMD | age-related macular degeneration |
| AMPK | AMP-activated protein kinase |
| ARD | age-related disease |
| ATG13 | autophagy related protein 13 |
| ATM | ataxia telangiectasia mutated |
| ATR | ATM-related |
| ATP | adenosine triphosphate |
| CMV | cytomegalovirus |
| CpG | 5’-C-p-G-3’ |
| CR | caloric restriction |
| DAP1 | death associated protein 1 |
| DN | diabetic neuropathy |
| eEF2 | eukaryotic elongation factor 2 |
| eIF | eukaryotic translation initiation factor |
| ER | endoplasmic reticulum |
| FA | fatty acid |
| FBW8 | F-Box And WD Repeat Domain Containing 8 |
| FDA | Food and Drug Administration |
| FKBP | FK506 binding protein |
| FK506 | Tacrolimus |
| FRB | FKBP12-Rapamycin Binding (FRB) domain of mTOR |
| GIT1 | GPCR-kinase interacting protein 1 |
| GSK3 | glycogen synthase kinase 3 |
| HD | Huntington’s disease |
| HGPS | Hutchinson Gilford progeroid syndrome |
| HIF1 | hypoxia inducible factor 1 |
| HTT | huntingtin protein |
| IC50 | half maximal inhibitory concentration |
| IKK | IkB kinase |
| IL | Interleukin |
| IGFR | insulin-like growth factor receptor |
| IMP2 | insulin-like growth factor 2 mRNA binding protein 2 |
| IRS | insulin receptor substrate |
| Ki | inhibitory constant |
| LAMTOR | late endosomal/lysosomal adaptor and MAPK and MTOR activator |
| LAP | liver-enriched activator protein |
| LARP1 | La-related protein |
| LIP | liver-enriched inhibitory protein |
| LKB1 | liver kinase B1 |
| LMNA | lamin A |
| L-DOPA | L-dopamine |
| MAPKAPK2 | mitogen-activated protein kinase-activated protein kinase 2 |
| MCF-7 | Michigan Cancer Foundation-7 (breast cancer cell line) |
| mLST8 | mammalian lethal with SEC13 protein 8 |
| MMP | matrix metalloproteinase |
| mTOR | mammalian/mechanistic target of rapamycin |
| mTORC1/2 | mTOR complex 1 or 2 |
| NFAT5 | nuclear factor of activated T cells 5 |
| OA | osteoarthritis |
| PD | Parkinson’s disease |
| PD-1 | programmed death 1 |
| PDK1/2 | pyruvate dehydrogenase kinase 1/2 |
| PGC-1-β | peroxisome proliferator-activated receptor gamma coactivator 1-β |
| PKC | protein kinase C |
| PPAR | Peroxisome Proliferator Activated Receptor |
| RA | rheumatoid arthritis |
| RAD | Ras associated with diabetes |
| RAGE | receptor for advanced glycation end products |
| REDD1 | regulated in development and DNA damage 1 |
| RNAi | RNA interference |
| ROS | reactive oxygen species |
| S6K | protein kinase that phosphorylates S6 ribosomal protein |
| SAβGAL | senescence associated beta galactosidase |
| SASP | senescence-associated secretory phenotype |
| SGK1 | serine/threonine protein kinase |
| SIRT | sirtuin |
| SOD1 | superoxide dismutase 1 |
| SREBP | sterol regulatory element-binding protein |
| STAT3 | signal transducer and activator of transcription 3 |
| T1DMT2DM | type 1 diabetes mellitustype 2 diabetes mellitus |
| TFEB | transcription factor EB |
| TNFα | tumour necrosis factor α |
| TSC1/2 | tuberous sclerosis complex 1 or 2 |
| ULK1 | Unc-51 like autophagy activating kinase |
| UTR | untranslated region |
| VEGF | vascular endothelial growth factor |
| γH2AX | Ser-139 phosphorylated histone 2A variant X |
References
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed cell senescence during mammalian embryonic development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef] [PubMed]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Zada, L.; Abou Karam, P.; Vadai, E.; Roitman, L.; Ovadya, Y.; Porat, Z.; Krizhanovsky, V. Quantitative identification of senescent cells in aging and disease. Aging Cell 2017, 16, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Herbig, U.; Ferreira, M.; Condel, L.; Carey, D.; Sedivy, J.M. Cellular senescence in aging primates. Science 2006, 311, 1257. [Google Scholar] [CrossRef] [PubMed]
- Smithson, L.J.; Gutmann, D.H. Proteomic analysis reveals GIT1 as a novel mTOR complex component critical for mediating astrocyte survival. Genes Dev. 2016, 30, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014, 36, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Ebner, M.; Sinkovics, B.; Szczygiel, M.; Ribeiro, D.W.; Yudushkin, I. Localization of mTORC2 activity inside cells. J. Cell Biol. 2017, 216, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Ortiz, D.; Academia, E.C.; Anies, A.C.; Liao, C.Y.; Kennedy, B.K. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell 2015, 14, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.J.; Wiese, H.; Tolle, R.C.; Zarei, M.; Dengjel, J.; Warscheid, B.; Thedieck, K. Functional Proteomics Identifies Acinus L as a Direct Insulin- and Amino Acid-Dependent Mammalian Target of Rapamycin Complex 1 (mTORC1) Substrate. Mol. Cell. Proteom. 2015, 14, 2042–2055. [Google Scholar] [CrossRef] [PubMed]
- Bandhakavi, S.; Kim, Y.M.; Ro, S.H.; Xie, H.; Onsongo, G.; Jun, C.B.; Kim, D.H.; Griffin, T.J. Quantitative nuclear proteomics identifies mTOR regulation of DNA damage response. Mol. Cell. Proteom. 2010, 9, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Smith, E.M.; Yelle, N.; Alain, T.; Bushell, M.; Pause, A. The ever-evolving role of mTOR in translation. Semin. Cell Dev. Biol. 2014, 36, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Powers, R.W., 3rd; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Muller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Levine, B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 2007, 3, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Taubert, S.; Crawford, D.; Libina, N.; Lee, S.J.; Kenyon, C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 2007, 6, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Gyurus, E.; Kaposztas, Z.; Kahan, B.D. Sirolimus therapy predisposes to new-onset diabetes mellitus after renal transplantation: A long-term analysis of various treatment regimens. Transpl. Proc. 2011, 43, 1583–1592. [Google Scholar] [CrossRef] [PubMed]
- Mizunuma, M.; Neumann-Haefelin, E.; Moroz, N.; Li, Y.; Blackwell, T.K. mTORC2-SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging Cell 2014, 13, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Nelson, G.; Rabanal-Ruiz, Y.; Kucheryavenko, O.; Dunhill-Turner, N.A.; Chesterman, C.C.; Zahari, Q.; Zhang, T.; Conduit, S.E.; Mitchell, C.A.; et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell Biol. 2017, 216, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Leontieva, O.V.; Blagosklonny, M.V. Gerosuppression by pan-mTOR inhibitors. Aging 2016, 8, 3535–3551. [Google Scholar] [CrossRef] [PubMed]
- Walters, H.E.; Deneka-Hannemann, S.; Cox, L.S. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging 2016, 8, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Makino, Y.; Okamoto, K.; Poellinger, L.; Ohnuma, K.; Morimoto, C.; Tanaka, H. TCR engagement increases hypoxia-inducible factor-1α protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J. Immunol. 2005, 174, 7592–7599. [Google Scholar] [CrossRef] [PubMed]
- Lerner, C.; Bitto, A.; Pulliam, D.; Nacarelli, T.; Konigsberg, M.; Van Remmen, H.; Torres, C.; Sell, C. Reduced mammalian target of rapamycin activity facilitates mitochondrial retrograde signaling and increases life span in normal human fibroblasts. Aging Cell 2013, 12, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Mendillo, M.L.; Tang, Y.C.; Subramanian, A.; Perley, C.C.; Roche, S.P.; Wong, B.; Narayan, R.; Kwon, H.; Koeva, M.; et al. Tight coordination of protein translation and HSF1 activation supports the anabolic malignant state. Science 2013, 341, 1238303. [Google Scholar] [CrossRef] [PubMed]
- Ortells, M.C.; Morancho, B.; Drews-Elger, K.; Viollet, B.; Laderoute, K.R.; Lopez-Rodriguez, C.; Aramburu, J. Transcriptional regulation of gene expression during osmotic stress responses by the mammalian target of rapamycin. Nucleic Acids Res. 2012, 40, 4368–4384. [Google Scholar] [CrossRef] [PubMed]
- Iadevaia, V.; Liu, R.; Proud, C.G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin. Cell Dev. Biol. 2014, 36, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Buchwalter, A.; Hetzer, M.W. Nucleolar expansion and elevated protein translation in premature aging. Nat. Commun. 2017, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W.; Spath, M.; Suchiman, H.E.D.; Muller, R.U.; Slagboom, P.E.; et al. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2016, 8, 16083. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Gygi, S.P.; Raught, B.; Polakiewicz, R.D.; Abraham, R.T.; Hoekstra, M.F.; Aebersold, R.; Sonenberg, N. Regulation of 4E-BP1 phosphorylation: A novel two-step mechanism. Genes Dev. 1999, 13, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Opdenaker, L.M.; Farach-Carson, M.C. Rapamycin selectively reduces the association of transcripts containing complex 5’ UTRs with ribosomes in C4-2B prostate cancer cells. J. Cell. Biochem. 2009, 107, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Zinzalla, V.; Stracka, D.; Oppliger, W.; Hall, M.N. Activation of mTORC2 by association with the ribosome. Cell 2011, 144, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Wu, C.C.; Kim, S.J.; Facchinetti, V.; Julien, L.A.; Finlan, M.; Roux, P.P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef] [PubMed]
- Takauji, Y.; Wada, T.; Takeda, A.; Kudo, I.; Miki, K.; Fujii, M.; Ayusawa, D. Restriction of protein synthesis abolishes senescence features at cellular and organismal levels. Sci. Rep. 2016, 6, 18722. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.B.; Zhang, X.; Sun, J.; Bennink, J.R.; Yewdell, J.W.; Patterson, C. mTORC1 links protein quality and quantity control by sensing chaperone availability. J. Biol. Chem. 2010, 285, 27385–27395. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Lamming, D.W. The Mechanistic Target of Rapamycin: The Grand ConducTOR of Metabolism and Aging. Cell Metab. 2016, 23, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Lee, K.Y.; Kim, J.R.; Choi, H.C. Interaction between mTOR pathway inhibition and autophagy induction attenuates adriamycin-induced vascular smooth muscle cell senescence through decreased expressions of p53/p21/p16. Exp. Gerontol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P.; et al. Autophagy is a critical regulator of memory CD8+ T cell formation. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J.J.; Jin, M.Y.; Gu, Y.T.; Wu, C.C.; Guo, W.J.; Yan, Y.Z.; Zhang, Z.J.; Wang, J.L.; Zhang, X.L.; et al. Sirt6 overexpression suppresses senescence and apoptosis of nucleus pulposus cells by inducing autophagy in a model of intervertebral disc degeneration. Cell Death Dis. 2018, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Kucharewicz, K.; Dudkowska, M.; Zawadzka, A.; Ogrodnik, M.; Szczepankiewicz, A.A.; Czarnocki, Z.; Sikora, E. Simultaneous induction and blockade of autophagy by a single agent. Cell Death Dis. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123 Pt 6, 917–926. [Google Scholar] [CrossRef]
- Rizza, S.; Cardaci, S.; Montagna, C.; Di Giacomo, G.; De Zio, D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A.; et al. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Niles, B.J.; Joslin, A.C.; Fresques, T.; Powers, T. TOR complex 2-Ypk1 signaling maintains sphingolipid homeostasis by sensing and regulating ROS accumulation. Cell Rep. 2014, 6, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Andres, D.A. mTORC2 is required for rit-mediated oxidative stress resistance. PLoS ONE 2014, 9, e115602. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Leontieva, O.V.; Blagosklonny, M.V. M(o)TOR of pseudo-hypoxic state in aging: Rapamycin to the rescue. Cell Cycle 2014, 13, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Alimbetov, D.; Davis, T.; Brook, A.J.; Cox, L.S.; Faragher, R.G.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Haidinger, M.; Katholnig, K.; Kopecky, C.; Poglitsch, M.; Lassnig, C.; Rosner, M.; Zlabinger, G.J.; Hengstschlager, M.; Muller, M.; et al. Inhibition of mTOR blocks the anti-inflammatory effects of glucocorticoids in myeloid immune cells. Blood 2011, 117, 4273–4283. [Google Scholar] [CrossRef] [PubMed]
- Saemann, M.D.; Haidinger, M.; Hecking, M.; Horl, W.H.; Weichhart, T. The multifunctional role of mTOR in innate immunity: Implications for transplant immunity. Am. J. Transpl. 2009, 9, 2655–2661. [Google Scholar] [CrossRef] [PubMed]
- Selvarajah, J.; Elia, A.; Carroll, V.A.; Moumen, A. DNA damage-induced S and G2/M cell cycle arrest requires mTORC2-dependent regulation of Chk1. Oncotarget 2015, 6, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK β1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Inoki, K.; Karbowniczek, M.; Petroulakis, E.; Sonenberg, N.; Henske, E.P.; Guan, K.L. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007, 26, 4812–4823. [Google Scholar] [CrossRef] [PubMed]
- Vadysirisack, D.D.; Ellisen, L.W. mTOR activity under hypoxia. Methods Mol. Biol. 2012, 821, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.P.; Leong, W.F.; Chau, J.F.; Jia, D.; Zeng, L.; Liu, H.; He, L.; Hao, A.; Zhang, H.; Meek, D.; et al. S6K1 is a multifaceted regulator of Mdm2 that connects nutrient status and DNA damage response. EMBO J. 2010, 29, 2994–3006. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.S.; Lane, D.P. Tumour suppressors, kinases and clamps: How p53 regulates the cell cycle in response to DNA damage. Bioessays 1995, 17, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; White, E. p53-dependent apoptosis pathways. Adv. Cancer Res. 2001, 82, 55–84. [Google Scholar] [PubMed]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT induces senescence in human cells via mTORC1 and p53 in the absence of DNA damage: Implications for targeting mTOR during malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Hu, H.; Tong, X.; Li, L.; Liu, X.; Chen, M.; Yuan, H.; Xie, X.; Li, Q.; Zhang, Y.; et al. The mTOR-S6K pathway links growth signalling to DNA damage response by targeting RNF168. Nat. Cell Biol. 2018, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, C.; Wang, X.; Briggs, M.R.; Admon, A.; Wu, J.; Hua, X.; Goldstein, J.L.; Brown, M.S. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 1993, 75, 187–197. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, P.G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brule, S.; Turcotte, V.; Cote, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Chinetti, G.; Fruchart, J.C.; Staels, B. Sorting out the roles of PPARα in energy metabolism and vascular homeostasis. J. Clin. Investig. 2006, 116, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Le Bacquer, O.; Petroulakis, E.; Paglialunga, S.; Poulin, F.; Richard, D.; Cianflone, K.; Sonenberg, N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J. Clin. Investig. 2007, 117, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Flor, A.C.; Wolfgeher, D.; Wu, D.; Kron, S.J. A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov. 2017, 3, 17075. [Google Scholar] [CrossRef] [PubMed]
- Vezina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, J.; Mahalati, K.; Kiberd, B.; McAlister, V.C.; MacDonald, A.S. Conversion to rapamycin immunosuppression in renal transplant recipients: Report of an initial experience. Transplantation 2000, 70, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Caron, E.; Ghosh, S.; Matsuoka, Y.; Ashton-Beaucage, D.; Therrien, M.; Lemieux, S.; Perreault, C.; Roux, P.P.; Kitano, H. A comprehensive map of the mTOR signaling network. Mol. Syst. Biol. 2010, 6, 453. [Google Scholar] [CrossRef] [PubMed]
- Stanfel, M.N.; Shamieh, L.S.; Kaeberlein, M.; Kennedy, B.K. The TOR pathway comes of age. Biochim. Biophys. Acta 2009, 1790, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Joung, S.M.; Park, Z.Y.; Rani, S.; Takeuchi, O.; Akira, S.; Lee, J.Y. Akt contributes to activation of the TRIF-dependent signaling pathways of TLRs by interacting with TANK-binding kinase 1. J. Immunol. 2011, 186, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Schickfluss, T.A.; Fattah, K.R.; Lee, K.J.; Chen, B.P.; Fehrenbacher, B.; Schaller, M.; Chen, D.J.; Rodemann, H.P. Function of erbB receptors and DNA-PKcs on phosphorylation of cytoplasmic and nuclear Akt at S473 induced by erbB1 ligand and ionizing radiation. Radiother. Oncol. 2011, 101, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [PubMed]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chene, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, Z.N.; Zubova, S.G.; Bukreeva, E.I.; Pospelov, V.A.; Pospelova, T.V.; Blagosklonny, M.V. Rapamycin decelerates cellular senescence. Cell Cycle 2009, 8, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Morris, M.; Hockey, H.-U.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Del Giudice, G.; Lattanzi, M.; Valiante, N.M.; Praestgaard, J.; Huang, B.; Lonetto, M.A.; Maecker, H.T.; Kovarik, J.; Carson, S.; et al. mTOR inhibition improves immune function in the elderly. Sci. Transl. Med. 2014, 6, 268ra179. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Aghajanian, C.; Raymond, E.; Olmos, D.; Schwartz, G.; Oelmann, E.; Grinsted, L.; Burke, W.; Taylor, R.; Kaye, S.; et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br. J. Cancer 2012, 107, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, S.V.; Gokhale, P.C.; Crew, A.P.; Cooke, A.; Yao, Y.; Mantis, C.; Kahler, J.; Workman, J.; Bittner, M.; Dudkin, L.; et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: Distinct from rapamycin. Mol. Cancer Ther. 2011, 10, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, A.; Khazaei, M.; Hasanzadeh, M.; ShahidSales, S.; Joudi Mashhad, M.; Farazestanian, M.; Sadeghnia, H.R.; Rezayi, M.; Maftouh, M.; Hassanian, S.M.; et al. Therapeutic Potential of Targeting PI3K/AKT Pathway in Treatment of Colorectal Cancer: Rational and Progress. J. Cell. Biochem. 2018, 119, 2460–2469. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Serrato, A.; Lee, J.; Holmes, B.; Landon, K.A.; Bashir, T.; Jung, M.E.; Lichtenstein, A.; Gera, J. Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma. PLoS ONE 2017, 12, e0176599. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Kurkjian, C.; Infante, J.R.; Bauer, T.M.; Burris, H.A., 3rd; Greco, F.A.; Shih, K.C.; Thompson, D.S.; Lane, C.M.; Finney, L.H.; et al. A phase 1 study of the sachet formulation of the oral dual PI3K/mTOR inhibitor BEZ235 given twice daily (BID) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Seront, E.; Rottey, S.; Filleul, B.; Glorieux, P.; Goeminne, J.C.; Verschaeve, V.; Vandenbulcke, J.M.; Sautois, B.; Boegner, P.; Gillain, A.; et al. Phase II study of dual phosphoinositol-3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor BEZ235 in patients with locally advanced or metastatic transitional cell carcinoma. BJU Int. 2016, 118, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Fazio, N.; Buzzoni, R.; Baudin, E.; Antonuzzo, L.; Hubner, R.A.; Lahner, H.; WW, D.E.H.; Raderer, M.; Teule, A.; Capdevila, J.; et al. A Phase II Study of BEZ235 in Patients with Everolimus-resistant, Advanced Pancreatic Neuroendocrine Tumours. Anticancer Res. 2016, 36, 713–719. [Google Scholar] [PubMed]
- Fan, Q.W.; Knight, Z.A.; Goldenberg, D.D.; Yu, W.; Mostov, K.E.; Stokoe, D.; Shokat, K.M.; Weiss, W.A. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006, 9, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Wu, Y.; Hou, W.; Sun, Z.; Wang, Y.; Wei, H.; Mo, W.; Yu, M. Bromodomain-containing protein 2 induces insulin resistance via the mTOR/Akt signaling pathway and an inflammatory response in adipose tissue. Cell. Signal. 2017, 30, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003, 52, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.S.; Jiang, N.N.; Zhou, Y.; Yu, K.Y.; Gong, H.Y.; Liao, G.J. LMO3 promotes gastric cancer cell invasion and proliferation through Akt-mTOR and Akt-GSK3β signaling. Int. J. Mol. Med. 2018, 41, 2755–2763. [Google Scholar] [CrossRef] [PubMed]
- Peng, N.; Meng, N.; Wang, S.; Zhao, F.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; Zhao, B.; Miao, J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E−/− mice. Sci. Rep. 2014, 4, 5519. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Han, L.; Huang, S.; Peng, N.; Wang, P.; Jiang, Z.; Zhao, J.; Su, L.; Zhang, S.; Zhang, Y.; et al. Identification of a novel MTOR activator and discovery of a competing endogenous RNA regulating autophagy in vascular endothelial cells. Autophagy 2014, 10, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Selleckchem.com. Available online: http://www.selleckchem.com/ (accessed on 3 June 2018).
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.E.; Burmeister, L.; Brooks, S.V.; Chan, C.C.; Friedline, S.; Harrison, D.E.; Hejtmancik, J.F.; Nadon, N.; Strong, R.; Wood, L.K.; et al. Rapamycin slows aging in mice. Aging Cell 2002, 11, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Pera, A.; Campos, C.; Lopez, N.; Hassouneh, F.; Alonso, C.; Tarazona, R.; Solana, R. Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas 2015, 82, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Hurez, V.; Dao, V.; Liu, A.; Pandeswara, S.; Gelfond, J.; Sun, L.; Bergman, M.; Orihuela, C.J.; Galvan, V.; Padron, A.; et al. Chronic mTOR inhibition in mice with rapamycin alters T, B, myeloid, and innate lymphoid cells and gut flora and prolongs life of immune-deficient mice. Aging Cell 2015, 14, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Capal, J.K.; Franz, D.N. Profile of everolimus in the treatment of tuberous sclerosis complex: An evidence-based review of its place in therapy. Neuropsychiatr. Dis. Treat. 2016, 12, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Caccamo, A.; Medina, D.X.; Benavides, A.D.; Javors, M.A.; Kraig, E.; Strong, R.; Richardson, A.; Oddo, S. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1β and enhancing NMDA signaling. Aging Cell 2012, 11, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Josselyn, S.A.; Frankland, P.W. mTORC2: Actin on your memory. Nat. Neurosci. 2013, 16, 379–380. [Google Scholar] [CrossRef] [PubMed]
- Thomanetz, V.; Angliker, N.; Cloetta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Ruegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Lang, U.E.; Heger, J.; Willbring, M.; Domula, M.; Matschke, K.; Tugtekin, S.M. Immunosuppression using the mammalian target of rapamycin (mTOR) inhibitor everolimus: Pilot study shows significant cognitive and affective improvement. Transpl. Proc. 2009, 41, 4285–4288. [Google Scholar] [CrossRef] [PubMed]
- An, W.L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.G.; Winblad, B.; Pei, J.J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Pei, J.J.; Hugon, J. mTOR-dependent signalling in Alzheimer’s disease. J. Cell. Mol. Med. 2008, 12, 2525–2532. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S. The role of mTOR signaling in Alzheimer disease. Front. Biosci. 2012, 4, 941–952. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-β, and Tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.L.; Zheng, W.; Halloran, J.J.; Burbank, R.R.; Hussong, S.A.; Hart, M.J.; Javors, M.; Shih, Y.Y.; Muir, E.; Solano Fonseca, R.; et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; De Pinto, V.; Messina, A.; Branca, C.; Oddo, S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J. Neurosci. 2014, 34, 7988–7998. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, L.; Farfara, D.; Shaham-Kol, M.; Vassar, R.; Frenkel, D.; Eldar-Finkelman, H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013, 288, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Monteys, A.M.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Biophys. Acta 2010, 1802, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, C.; Chen, S.; Ye, Y.; Guo, M.; Ren, Q.; Liu, L.; Zhang, H.; Xu, C.; Zhou, Q.; et al. Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell. Signal. 2014, 26, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S.; et al. Rotenone induction of hydrogen peroxide inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E pathways, leading to neuronal apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.P.; Chen, J.; Zhao, Y.; Chai, Z.; Hu, Y. mTOR Signaling in Parkinson’s Disease. Neuromol. Med. 2017, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [PubMed]
- Kolosova, N.G.; Muraleva, N.A.; Zhdankina, A.A.; Stefanova, N.A.; Fursova, A.Z.; Blagosklonny, M.V. Prevention of age-related macular degeneration-like retinopathy by rapamycin in rats. Am. J. Pathol. 2012, 181, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T.; Dresner, S.; Forooghian, F.; Glaser, T.; Doss, L.; Zhou, M.; Cunningham, D.; Shimel, K.; Harrington, M.; Hammel, K.; et al. Treatment of geographic atrophy with subconjunctival sirolimus: Results of a phase I/II clinical trial. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell-derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Kawakami, Y.; Lavasani, M.; Mu, X.; Cummins, J.H.; Yurube, T.; Kuroda, R.; Kurosaka, M.; Fu, F.H.; Robbins, P.D.; et al. mTOR signaling plays a critical role in the defects observed in muscle-derived stem/progenitor cells isolated from a murine model of accelerated aging. J. Orthop. Res. 2017, 35, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Graziotto, J.J.; Blair, C.D.; Mazzulli, J.R.; Erdos, M.R.; Krainc, D.; Collins, F.S. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci. Transl. Med. 2011, 3, 89ra58. [Google Scholar] [CrossRef] [PubMed]
- Progeria Research Foundation (PRF). Available online: https://www.progeriaresearch.org/clinical-trials/ (accessed on 3 June 2018).
- Choi, J.C.; Wu, W.; Muchir, A.; Iwata, S.; Homma, S.; Worman, H.J. Dual specificity phosphatase 4 mediates cardiomyopathy caused by lamin A/C (LMNA) gene mutation. J. Biol. Chem. 2012, 287, 40513–40524. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.J.; Chen, S.C.; Garelick, M.G.; Dai, D.F.; Liao, C.Y.; Schreiber, K.H.; MacKay, V.L.; An, E.H.; Strong, R.; Ladiges, W.C.; et al. Rapamycin reverses elevated mTORC1 signaling in lamin A/C-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci. Transl. Med. 2012, 4, 144ra103. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Kennedy, B.K. When lamins go bad: Nuclear structure and disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, B.; Shimokata, T.; Honda, K.; Kondoh, C.; Hayashi, N.; Yoshino, Y.; Sassa, N.; Nakano, Y.; Gotoh, M.; Ando, Y. Muscle wasting associated with the long-term use of mTOR inhibitors. Mol. Clin. Oncol. 2016, 5, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Urfer, S.R.; Kaeberlein, T.L.; Mailheau, S.; Bergman, P.J.; Creevy, K.E.; Promislow, D.E.L.; Kaeberlein, M. A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. Geroscience 2017, 39, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Naumann, D.N.; Toman, E.; Davies, D.; Bishop, J.R.B.; Su, Z.; Hampson, P.; Dinsdale, R.J.; Crombie, N.; Duggal, N.A.; et al. Prehospital immune responses and development of multiple organ dysfunction syndrome following traumatic injury: A prospective cohort study. PLoS Med. 2017, 14, e1002338. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Jackson, T.; Sapey, E.; Lord, J.M. Frailty and sarcopenia: The potential role of an aged immune system. Ageing Res. Rev. 2017, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Smink, J.J.; Begay, V.; Schoenmaker, T.; Sterneck, E.; de Vries, T.J.; Leutz, A. Transcription factor C/EBPβ isoform ratio regulates osteoclastogenesis through MafB. EMBO J. 2009, 28, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Smink, J.J.; Leutz, A. Rapamycin and the transcription factor C/EBPβ as a switch in osteoclast differentiation: Implications for lytic bone diseases. J. Mol. Med. 2010, 88, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Glantschnig, H.; Fisher, J.E.; Wesolowski, G.; Rodan, G.A.; Reszka, A.A. M-CSF, TNFα and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell Death Differ. 2003, 10, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Kneissel, M.; Luong-Nguyen, N.H.; Baptist, M.; Cortesi, R.; Zumstein-Mecker, S.; Kossida, S.; O’Reilly, T.; Lane, H.; Susa, M. Everolimus suppresses cancellous bone loss, bone resorption, and cathepsin K expression by osteoclasts. Bone 2004, 35, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Cejka, D.; Hayer, S.; Niederreiter, B.; Sieghart, W.; Fuereder, T.; Zwerina, J.; Schett, G. Mammalian target of rapamycin signaling is crucial for joint destruction in experimental arthritis and is activated in osteoclasts from patients with rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Bruyn, G.A.; Tate, G.; Caeiro, F.; Maldonado-Cocco, J.; Westhovens, R.; Tannenbaum, H.; Bell, M.; Forre, O.; Bjorneboe, O.; Tak, P.P.; et al. Everolimus in patients with rheumatoid arthritis receiving concomitant methotrexate: A 3-month, double-blind, randomised, placebo-controlled, parallel-group, proof-of-concept study. Ann. Rheum. Dis. 2008, 67, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Pal, B.; Endisha, H.; Zhang, Y.; Kapoor, M. mTOR: A potential therapeutic target in osteoarthritis? Drugs R&D 2015, 15, 27–36. [Google Scholar] [CrossRef]
- Lecka-Czernik, B. Diabetes, bone and glucose-lowering agents: Basic biology. Diabetologia 2017, 60, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Creecy, A.; Uppuganti, S.; Merkel, A.R.; O’Neal, D.; Makowski, A.J.; Granke, M.; Voziyan, P.; Nyman, J.S. Changes in the Fracture Resistance of Bone with the Progression of Type 2 Diabetes in the ZDSD Rat. Calcif. Tissue Int. 2016, 99, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Mera, P.; Laue, K.; Ferron, M.; Confavreux, C.; Wei, J.; Galan-Diez, M.; Lacampagne, A.; Mitchell, S.J.; Mattison, J.A.; Chen, Y.; et al. Osteocalcin Signaling in Myofibers Is Necessary and Sufficient for Optimum Adaptation to Exercise. Cell Metab. 2016, 23, 1078–1092. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Brooks, R.; Houskeeper, J.; Bremner, S.K.; Dunlop, J.; Viollet, B.; Logan, P.J.; Salt, I.P.; Ahmed, S.F.; Yarwood, S.J. Corrigendum to “Metformin suppresses adipogenesis through both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms” [Mol. Cell. Endocrinol. 440 15 January 2017 57–68]. Mol. Cell. Endocrinol. 2017, 443, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tang, Y.; Jin, X.; Chen, C.; Lu, Y.; Liu, L.; Shen, C. Metformin Inhibits Advanced Glycation End Products-Induced Inflammatory Response in Murine Macrophages Partly through AMPK Activation and RAGE/NFkappaB Pathway Suppression. J. Diabetes Res. 2016, 2016, 4847812. [Google Scholar] [CrossRef] [PubMed]
- Shioi, T.; McMullen, J.R.; Tarnavski, O.; Converso, K.; Sherwood, M.C.; Manning, W.J.; Izumo, S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 2003, 107, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- McMullen, J.R.; Sherwood, M.C.; Tarnavski, O.; Zhang, L.; Dorfman, A.L.; Shioi, T.; Izumo, S. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation 2004, 109, 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, D.M.; Singh, R.S.; Franklin, D.P.; Carey, D.J.; Elmore, J.R. Rapamycin suppresses experimental aortic aneurysm growth. J. Vasc. Surg. 2004, 40, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Flynn, J.M.; O’Leary, M.N.; Zambataro, C.A.; Academia, E.C.; Presley, M.P.; Garrett, B.J.; Zykovich, A.; Mooney, S.D.; Strong, R.; Rosen, C.J.; et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell 2013, 12, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhu, X.; Xie, W.; Han, P.; Li, K.; Sun, Z.; Wang, Y.; Chen, C.; Song, R.; Cao, C.; et al. Rad as a novel regulator of excitation-contraction coupling and β-adrenergic signaling in heart. Circ. Res. 2010, 106, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Karunadharma, P.P.; Chiao, Y.A.; Basisty, N.; Crispin, D.; Hsieh, E.J.; Chen, T.; Gu, H.; Djukovic, D.; Raftery, D.; et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 2014, 13, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Salloum, F.; Das, A.; Xi, L.; Vetrovec, G.W.; Kukreja, R.C. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 41, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Salloum, F.N.; Durrant, D.; Ockaili, R.; Kukreja, R.C. Rapamycin protects against myocardial ischemia-reperfusion injury through JAK2-STAT3 signaling pathway. J. Mol. Cell. Cardiol. 2012, 53, 858–869. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Salloum, F.N.; Filippone, S.M.; Durrant, D.E.; Rokosh, G.; Bolli, R.; Kukreja, R.C. Inhibition of mammalian target of rapamycin protects against reperfusion injury in diabetic heart through STAT3 signaling. Basic Res. Cardiol. 2015, 110, 31. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Durrant, D.; Koka, S.; Salloum, F.N.; Xi, L.; Kukreja, R.C. Mammalian target of rapamycin (mTOR) inhibition with rapamycin improves cardiac function in type 2 diabetic mice: Potential role of attenuated oxidative stress and altered contractile protein expression. J. Biol. Chem. 2014, 289, 4145–4160. [Google Scholar] [CrossRef] [PubMed]
- Morice, M.C.; Serruys, P.W.; Sousa, J.E.; Fajadet, J.; Ban Hayashi, E.; Perin, M.; Colombo, A.; Schuler, G.; Barragan, P.; Guagliumi, G.; et al. A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization. N. Engl. J. Med. 2002, 346, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Kim, J.; Schrier, R.W.; Edelstein, C.L. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.R.; Serra, A.L.; Le Hir, M.; Molle, K.D.; Hall, M.N.; Wuthrich, R.P. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transpl. 2006, 21, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Zafar, I.; Belibi, F.A.; He, Z.; Edelstein, C.L. Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD). Nephrol Dial. Transpl. 2009, 24, 2349–2353. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Peces, R.; Peces, C.; Perez-Duenas, V.; Cuesta-Lopez, E.; Azorin, S.; Selgas, R. Rapamycin reduces kidney volume and delays the loss of renal function in a patient with autosomal-dominant polycystic kidney disease. NDT Plus 2009, 2, 133–135. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Du, H.; King, B.F.; Kumar, S.; Dean, P.G.; Cosio, F.G.; Torres, V.E. Sirolimus reduces polycystic liver volume in ADPKD patients. J. Am. Soc. Nephrol. 2008, 19, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Shao, Y.Q.; He, Q. Sirolimus for treatment of autosomal-dominant polycystic kidney disease: A meta-analysis of randomized controlled trials. Transpl. Proc. 2014, 46, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Myint, T.M.; Rangan, G.K.; Webster, A.C. Treatments to slow progression of autosomal dominant polycystic kidney disease: Systematic review and meta-analysis of randomized trials. Nephrology 2014, 19, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Bolignano, D.; Palmer, S.C.; Ruospo, M.; Zoccali, C.; Craig, J.C.; Strippoli, G.F. Interventions for preventing the progression of autosomal dominant polycystic kidney disease. Cochrane Database Syst. Rev. 2015, CD010294. [Google Scholar] [CrossRef] [PubMed]
- Jardine, M.J.; Liyanage, T.; Buxton, E.; Perkovic, V. mTOR inhibition in autosomal-dominant polycystic kidney disease (ADPKD): The question remains open. Nephrol. Dial. Transpl. 2013, 28, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, G.; Schieppati, A.; Ruggenenti, P. Clinical practice. Nephropathy in patients with type 2 diabetes. N. Engl. J. Med. 2002, 346, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Ruegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [PubMed]
- Lloberas, N.; Cruzado, J.M.; Franquesa, M.; Herrero-Fresneda, I.; Torras, J.; Alperovich, G.; Rama, I.; Vidal, A.; Grinyo, J.M. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J. Am. Soc. Nephrol. 2006, 17, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, J.; Qin, L.; Shou, Z.; Zhao, J.; Wang, H.; Chen, Y.; Chen, J. Rapamycin prevents early steps of the development of diabetic nephropathy in rats. Am. J. Nephrol. 2007, 27, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, M.; Isono, M.; Isshiki, K.; Sugimoto, T.; Koya, D.; Kashiwagi, A. Inhibition of mTOR signaling with rapamycin attenuates renal hypertrophy in the early diabetic mice. Biochem. Biophys. Res. Commun. 2006, 340, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Inoki, K.; Masutani, K.; Wakabayashi, Y.; Komai, K.; Nakagawa, R.; Guan, K.L.; Yoshimura, A. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem. Biophys. Res. Commun. 2009, 384, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Chen, J.; Mao, X. Everolimus vs. rapamycin for treating diabetic nephropathy in diabetic mouse model. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Kenerson, H.L.; Subramanian, S.; McIntyre, R.; Kazami, M.; Yeung, R.S. Livers with constitutive mTORC1 activity resist steatosis independent of feedback suppression of Akt. PLoS ONE 2015, 10, e0117000. [Google Scholar] [CrossRef] [PubMed]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Hornsby, P.J. Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [PubMed]
- Alayev, A.; Salamon, R.S.; Schwartz, N.S.; Berman, A.Y.; Wiener, S.L.; Holz, M.K. Combination of Rapamycin and Resveratrol for Treatment of Bladder Cancer. J. Cell. Physiol. 2017, 232, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Dilly, A.K.; Kim, S.Y.; Choudry, H.A.; Lee, Y.J. Rapamycin-enhanced mitomycin C-induced apoptotic death is mediated through the S6K1-Bad-Bak pathway in peritoneal carcinomatosis. Cell Death Dis. 2014, 5, e1281. [Google Scholar] [CrossRef] [PubMed]
- Sendur, M.A.; Zengin, N.; Aksoy, S.; Altundag, K. Everolimus: A new hope for patients with breast cancer. Curr. Med. Res. Opin. 2014, 30, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, J.; van den Bogaard, J.; Jutten, B.; Belgers, E.; Sosef, M.; Leijtens, J.W.; Beets, G.L.; Jansen, R.L.; Riedl, R.G.; Clarijs, R.; et al. A phase I-II study on the combination of rapamycin and short course radiotherapy in rectal cancer. Radiother. Oncol. 2015, 116, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Bennani, N.N.; LaPlant, B.R.; Ansell, S.M.; Habermann, T.M.; Inwards, D.J.; Micallef, I.N.; Johnston, P.B.; Porrata, L.F.; Colgan, J.P.; Markovic, S.N.; et al. Efficacy of the oral mTORC1 inhibitor everolimus in relapsed or refractory indolent lymphoma. Am. J. Hematol. 2017, 92, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.B.; Pinter-Brown, L.C.; Warsi, G.; White, K.; Ramchandren, R. Phase 2 study of everolimus for relapsed or refractory classical Hodgkin lymphoma. Exp. Hematol. Oncol. 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Hodis, E.; Jacobus, S.; Supko, J.G.; Stewart, M.; Choueiri, T.K.; Gandhi, L.; Cleary, J.M.; et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 2014, 4, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.B.; Lee, H.Y.; Young, D.M.; Tien, A.C.; Rowson-Baldwin, A.; Shu, Y.Y.; Jan, Y.N.; Jan, L.Y. Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J. Mol. Med. 2012, 90, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, M.; Ketzinel-Gilad, M.; Ariav, Y.; Pappo, O.; Karaca, M.; Castel, J.; Berthault, M.F.; Magnan, C.; Cerasi, E.; Kaiser, N.; et al. mTOR inhibition by rapamycin prevents β-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 2008, 57, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Arriola Apelo, S.I.; Neuman, J.C.; Baar, E.L.; Syed, F.A.; Cummings, N.E.; Brar, H.K.; Pumper, C.P.; Kimple, M.E.; Lamming, D.W. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 2016, 15, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Fernandez, E.; Liu, Y.; Strong, R.; Salmon, A.B. Metformin reduces glucose intolerance caused by rapamycin treatment in genetically heterogeneous female mice. Aging 2018. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.A.; Travers, J.B.; Machado, C.; Somani, A.K.; Spandau, D.F. Reversing the aging stromal phenotype prevents carcinoma initiation. Aging 2011, 3, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Dore, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Contribution to Complex | mTORC1 | mTORC2 |
|---|---|---|
| core | mTOR | mTOR |
| mLST8/Gβ3 | mLST8/Gβ3 | |
| Deptor | Deptor | |
| Tti1/Tel2 | Tti1/Tel2 | |
| complex-specific | Raptor | Rictor |
| PRAS40 | ||
| mSIN1 | ||
| Protor1/2 |
| mTORC1 | mTORC2 | |
|---|---|---|
| localization when active | lysosome | ribosome, plasma membrane, mitochondria, endoplasmic reticulum, lysosome |
| targets activated | S6KT389, HIF 1α, GSK3, SOD1, Grb10, eIF4G, Acinus L, eEF2, IMP2 | SGK1, PKC, paxillin, Rho GTPases, AktS473, IGFR, PDK1 |
| targets inhibited | 4EBP1/2, Maf1, Lipin-1, ULK1, ATG13, TFEB, DAP1, LARP1 | FBW8 |
| activated by | insulin, growth factors, Rheb, Rag, Akt, amino acids, high O2, cytokines, TNFα, IkkB | PI3K, growth factors including IGFR, Akt (on mSIN1), membrane tension, ROS, ATM/ATR |
| inhibited by | AMPK, TSC1/2 (via Rheb inactivation), low O2, low ATP, low amino acids | S6K on both Rictor and mSIN1 TSC1/2 (via Rheb inactivation) |
| biochemical outcomes of activation | protein, nucleotide, lipid and mitochondrial biosynthesis; inhibition of autophagy | actin reorganization, lipid biosynthesis |
| overall outcomes of activation | cell growth (increase in volume and biomass) cell proliferation suppression of oxidative damage | cell size (surface area increase) cell shape (cytoskeletal changes) survival under oxidative stresscell cycle progression metabolic control |
| Drug Class | Mode of Action | Drug Name | Ki or IC50 | Status |
|---|---|---|---|---|
| mTORC1 inhibitor | Binds FKBP12 which then associates with mTORC1 and partially occludes kinase active site; mTORC2 inhibited on chronic treatment (possibly through feedback loops) | Rapamycin (sirolimus) | mTORC1 IC50 0.1 nM (in HEK293 cells) | FDA-approved for cancer and as immunosuppressant to prevent rejection in renal transplant; eluting stents in cardiovascular disease Delays senescence in cell culture [109]; extends lifespan and health in lab animals and improves cardiovascular health in companion dogs (see text) |
| Everolimus (RAD001) | mTORC1 IC50 1.6–2.4 nM (cell-free assay) | FDA-approved for cancer (e.g., monotherapy against advanced renal cell carcinoma, neuroendocrine tumours of pancreatic, gastrointestinal or lung origin, and SEGA associated with TSC, and as combination therapy with exemestane for HER2-negative breast cancer). Clinical trials show immune system rejuvenation [110,111] | ||
| Temsirolimus; (CCI-779, NSC 683864) | IC50 0.3–0.5 nM in cell culture | FDA approved, used at 10 mg/kg/day in acute lymphocytic leukaemia | ||
| Pan-mTOR inhibitor (inhibits both mTORC1 and mTORC2) | ATP-competitive mTORC1/2 inhibitor | AZD8055 | mTOR IC50 0.8 nM (MDA-MB-468 cells); 1000-fold selectivity against PI3K isoforms and ATM/DNA-PK | Acceptable safety profile for treatment of advanced solid tumours and lymphoma in phase I trial [112]; reverses phenotypes of senescence in cell culture [34] |
| Sapanisertib (AK-228, INK 128, MLN0128) | mTORC1 and mTORC2 1 nM (PI3K isoforms ~200 nM) | Phase 1 trials (cancer) | ||
| OSI-027 | 22 nM mTORC1, 65 nM mTORC2 (>100× selectivity over PI3K) | Phase 1 trials; in experimental colorectal xenograft, OSI-027 (65 mg/kg) more effective than rapamycin [113], reviewed [114] | ||
| mTORC2-specific inhibitor | Prevents interaction of Rictor with mTOR hence blocking mTORC2 | JR-AB2 | Experimental, xenograft tumour models [115] | |
| Dual PI3K and mTOR inhibitor | ATP-competitive dual PI3K and mTORC1/2 inhibitor | Apitolisib (GDC-0980, RG7422) | Dual PI3K/mTOR 5–14 nM Ki, 17 nM mTOR | Phase 2 trials (cancer) |
| Dactolisib (NVP-BEZ235, BEZ235) | mTOR IC50 6 nM, PI3K p110α/γ/δ IC50 4/5/7 nM respectively; IC50 ATR 21 nM (cell-free assays) | Passed phase I initial dose discovery trial [116]; modest efficacy in advanced or metastatic carcinoma in phase II [117] but poorly tolerated in advanced pancreatic neuroendocrine tumour patient phase II study [118]; beneficial outcomes in trial with everolimus for reversal of immune senescence [110] | ||
| PF-04691502 | PI3K(α/β/δ/γ)/mTOR dual inhibitor with Ki of 1.8/2.1/1.6/1.9 and 16 nM (respectively) | Phase 1 clinical trials | ||
| PI3K, DNAPK and mTOR | ATP binding site competitor | PI-103 | PI3K 2–15 nM, mTOR and DNAPK 30 nM | Experimental [119] |
| Other components of signalling pathway | PI3K and BRD bromodomain proteins | SF2523 | DNAPK 9, 34–158 nM; BRD4 241 nM, mTOR 280 nM | Blocks Brd4; blocks Brd2 to overcome insulin resistance—may be useful as adjunct to prevent diabetic complications of mTOR inhibitors [120] |
| Highly selective GSK3 inhibitor; ATP binding competitor | CHIR-98014 | GSK3α 0.65 nM GSK3β 0.58 nM | Experimental [121,122] | |
| mTOR activator | FKBP1A | 3BDO | N/A | Experimental; inhibits autophagy; provides vascular protection [123]; improves neuronal function in App and Psen1 transgenic mice [124] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walters, H.E.; Cox, L.S. mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2325. https://doi.org/10.3390/ijms19082325
Walters HE, Cox LS. mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. International Journal of Molecular Sciences. 2018; 19(8):2325. https://doi.org/10.3390/ijms19082325
Chicago/Turabian StyleWalters, Hannah E., and Lynne S. Cox. 2018. "mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases" International Journal of Molecular Sciences 19, no. 8: 2325. https://doi.org/10.3390/ijms19082325
APA StyleWalters, H. E., & Cox, L. S. (2018). mTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. International Journal of Molecular Sciences, 19(8), 2325. https://doi.org/10.3390/ijms19082325