Phylogenomic and Comparative Analyses of Complete Plastomes of Croomia and Stemona (Stemonaceae)

Abstract
:1. Introduction
2. Results and Discussion
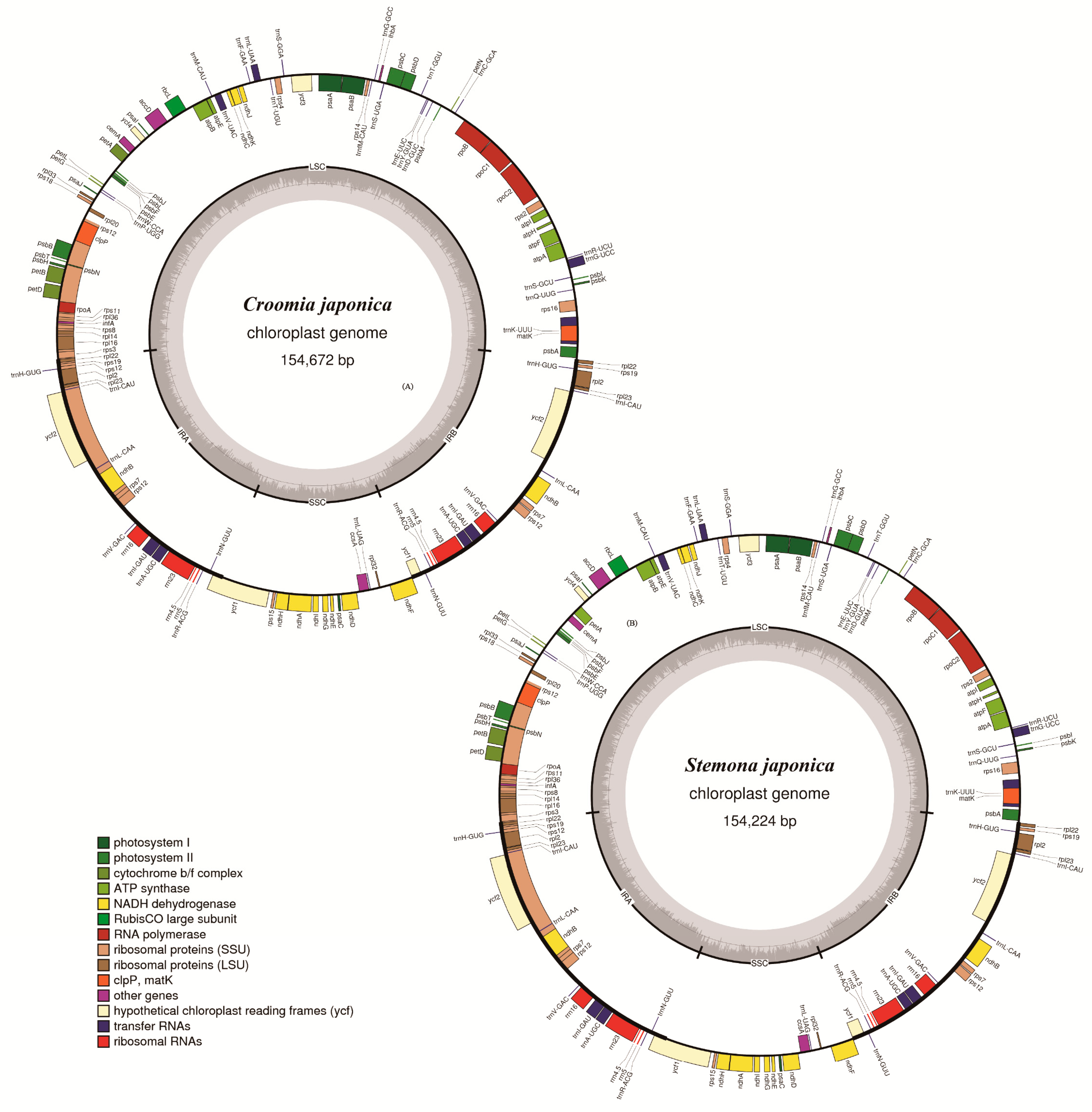
2.1. Genome Assembly and Features
2.2. Contraction and Expansion of Inverted Repeats
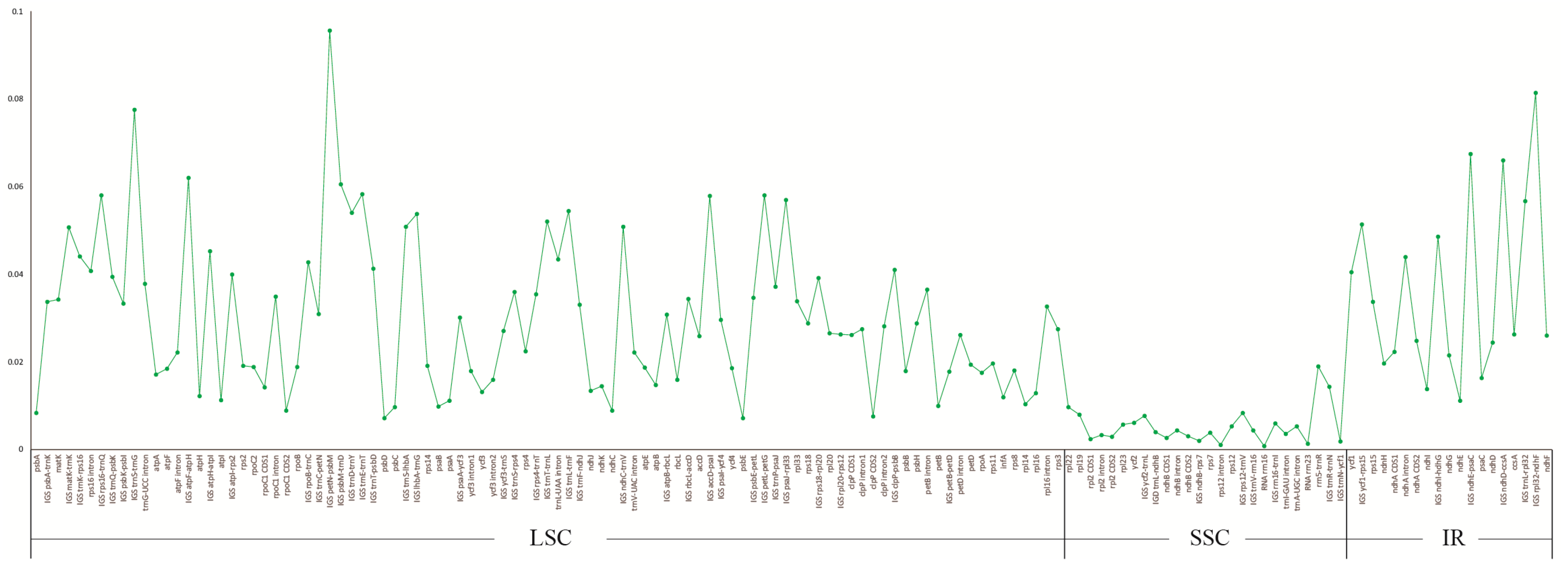
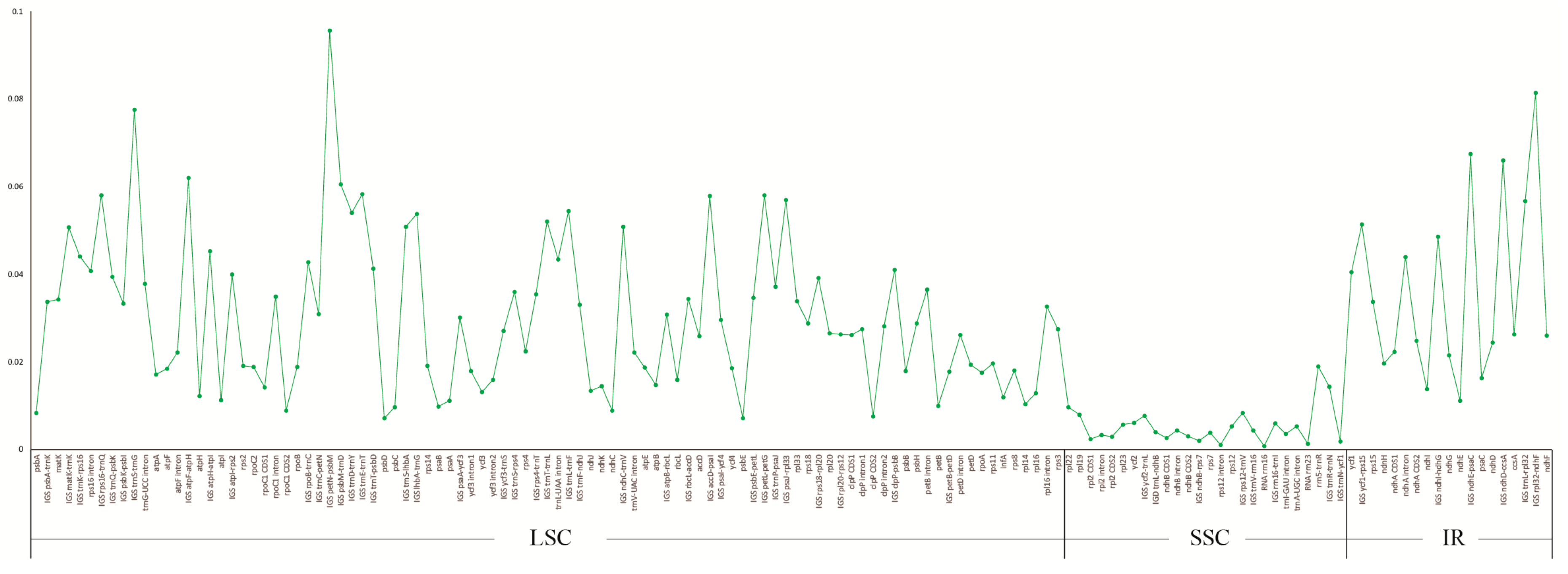
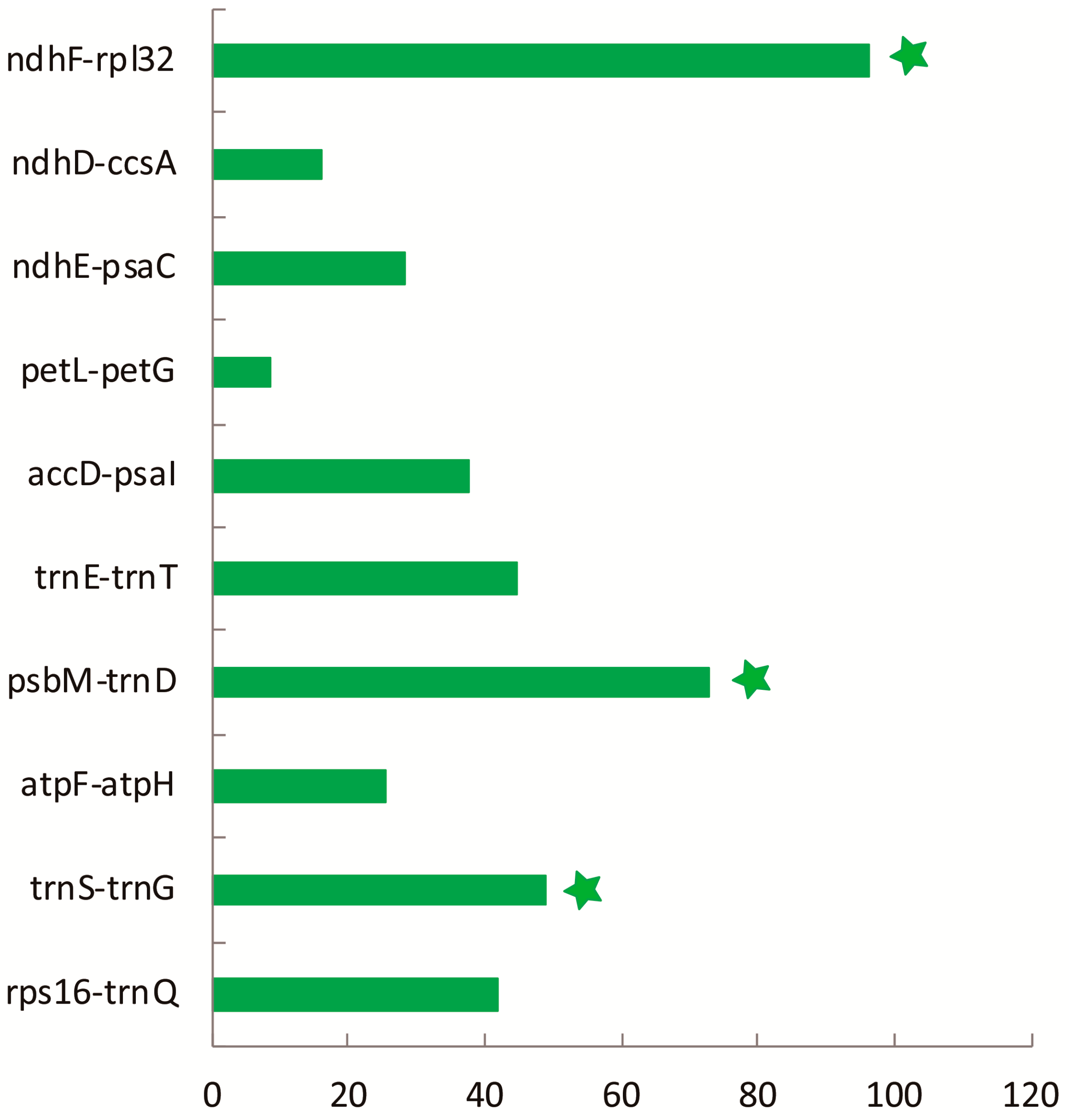
2.3. Divergence Hotspot Regions
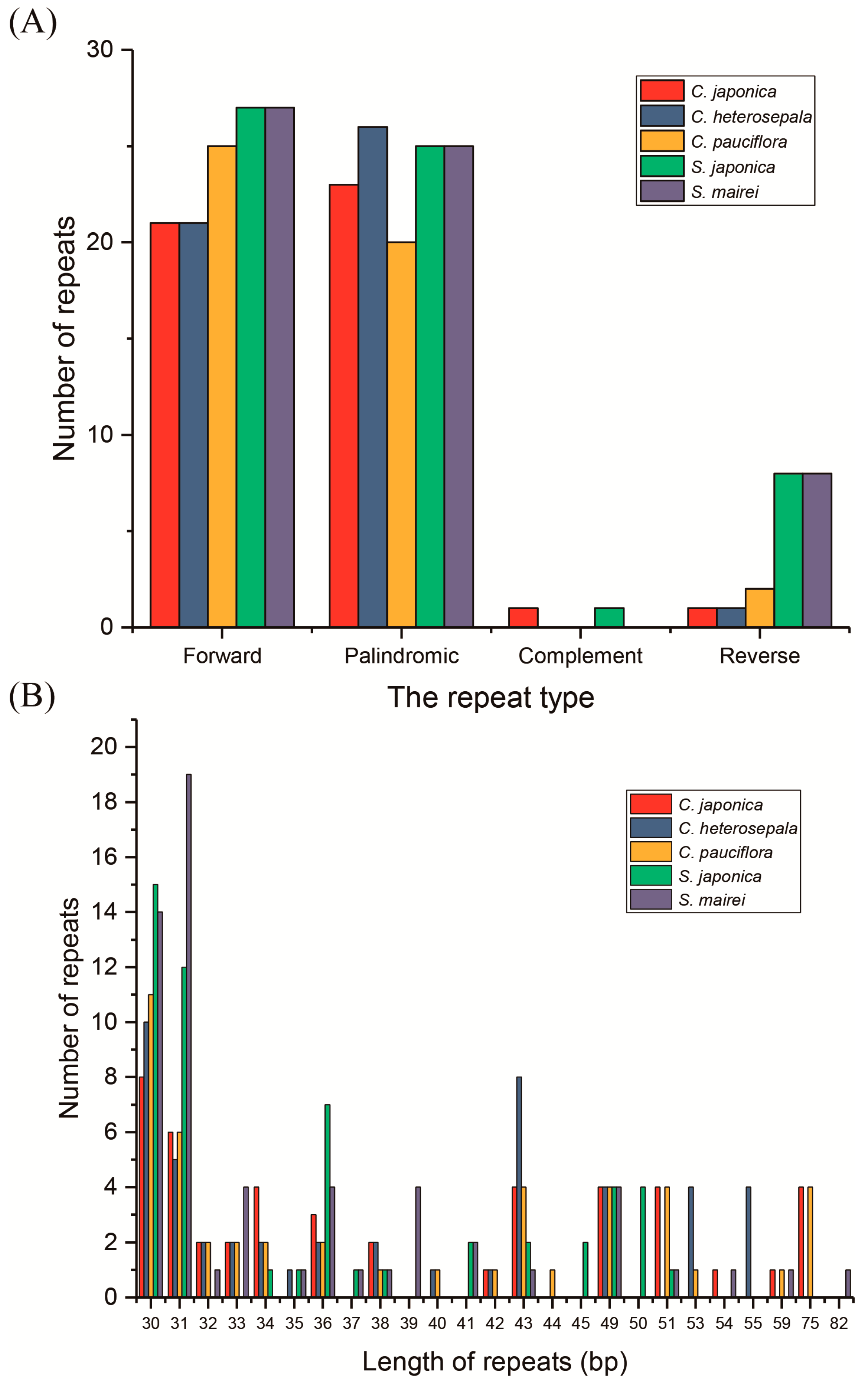
2.4. Repetitive Sequences and SSR Polymorphisms
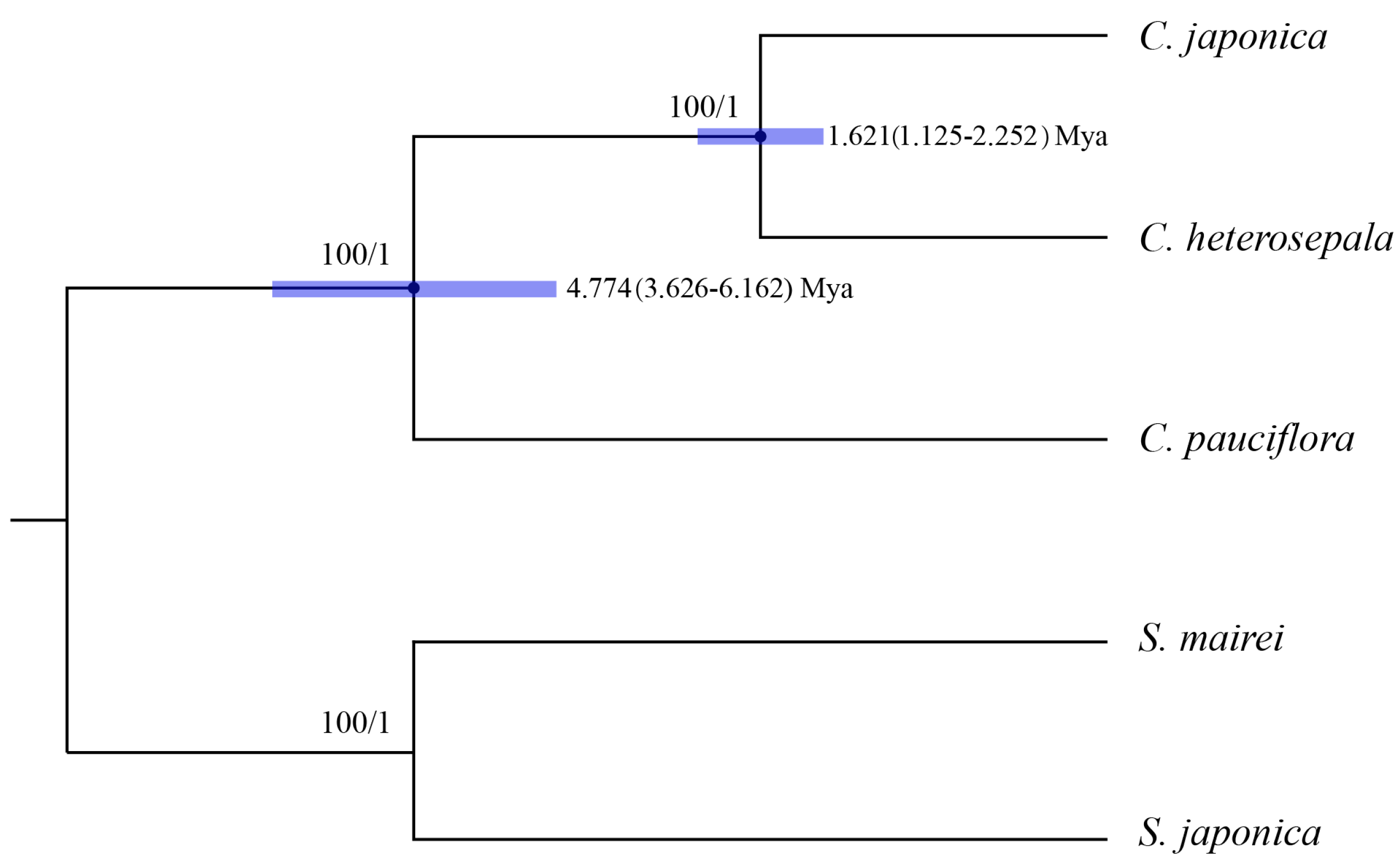
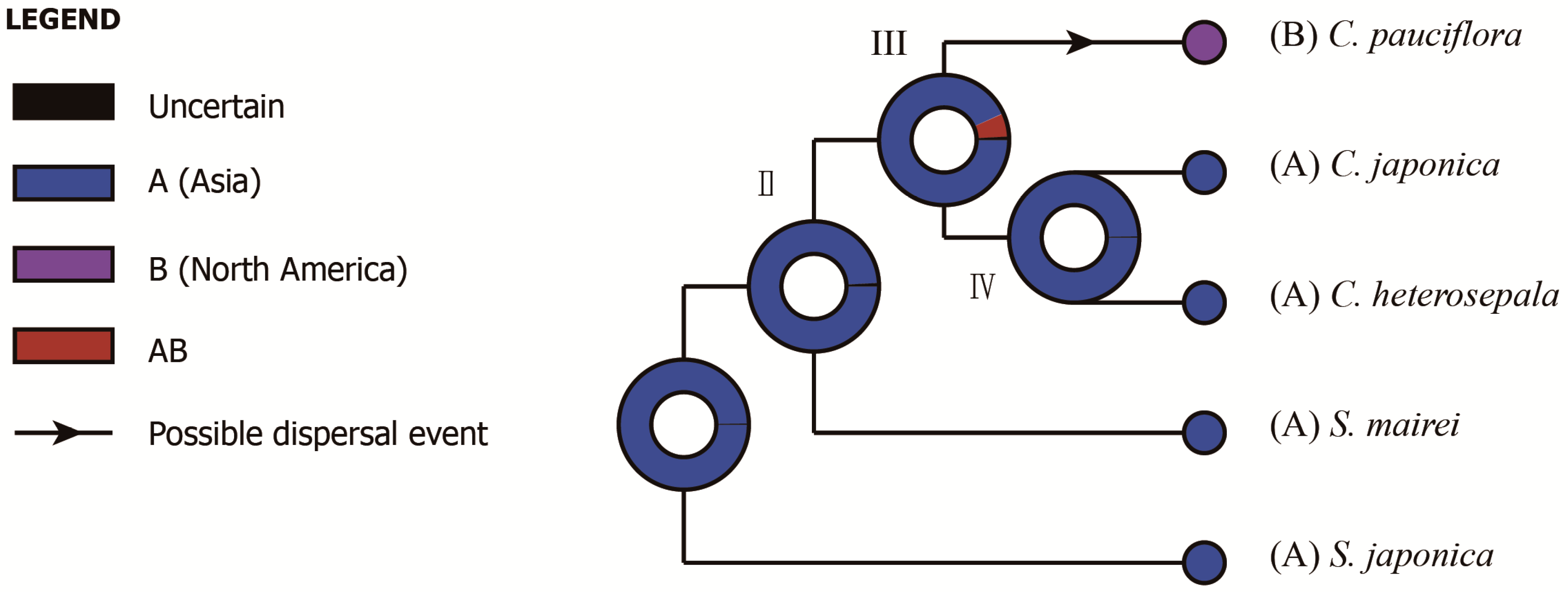
2.5. Phylogenetic Analysis, Divergence Time and Ancestral Area Reconstruction
3. Materials and Methods
3.1. Sample Preparation, Sequencing, Assembly and Validation
3.2. Genome Annotation and Whole Genome Comparison
3.3. Characterization of Repeat Sequence and SSRs
3.4. Phylogenetic Analysis, Divergence Time and Ancestral Area Reconstruction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| cp | Complete chloroplast |
| IR | Inverted Repeat |
| lSC | Large Single Copy |
| SSC | Small Single Copy |
| Pi | Nucleotide variability |
| SSR | Simple Sequence Repeat |
| PIC | Potentially Informative Character |
| ML | Maximum likelihood |
| BI | Bayesian Inference |
| MCMC | Markov chain Monte Carlo |
Appendix A


References
- Rogers, G.K. The Stemonaceae in the southeastern United States. J. Arnold Arboretum 1982, 63, 327–336. [Google Scholar]
- Whetstone, R.D. Notes on Croomia pauciflora (Stemonaceae). Rhodora 1984, 25, 131–137. [Google Scholar]
- Li, E.X. Studies on Phylogeography of Croomia and Phylogeny of Croomia and Its Allies. Ph.D. Thesis, Zhejiang University, Hangzhou, China, 2006. [Google Scholar]
- Okuyama, S. On the Japanese species of Croomia. J. Jpn. Bot. 1944, 20, 31–32. [Google Scholar]
- Ohwi, J. Croomia. In Flora of Japan; Smithsonian Institution: Washington, DC, USA, 1965; p. 279. [Google Scholar]
- Lin, W.; Cai, M.; Ying, B.; Feng, R. Studies on the chemical constituents of Croomia japonica Miq. Yao Xue Xue Bao 1993, 28, 202–206. [Google Scholar] [PubMed]
- Pilli, R.A.; Ferreira, M.C. Recent progress in the chemistry of the Stemona alkaloids. Nat. Prod. Rep. 2000, 17, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.H.; Duyfjes, B.E. “Stemonaceae”, in Flora of China, Flagellariaceae through Marantaceae. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; Missouri Botanical Garden Press: Beijing, China, 2000; Volume 24, pp. 70–72. [Google Scholar]
- Patrick, T.S.; Allison, J.; Krakow, G. Protected plants of Georgia. Georgia Department of Natural Resources, Natural Heritage Program. Soc. Circ. 1995, 25, 36–38. [Google Scholar]
- Estill, J.C.; Cruzan, M.B. Phytogeography of rare plant species endemic to the southeastern United States. Castanea 2001, 36, 3–23. [Google Scholar]
- Sung, W.; Yan, X. China Species Red List; Higher Education Press: Beijing, China, 2004. [Google Scholar]
- Li, E.; Yi, S.; Qiu, Y.; Guo, J.; Comes, H.P.; Fu, C. Phylogeography of two East Asian species in Croomia (Stemonaceae) inferred from chloroplast DNA and ISSR fingerprinting variation. Mol. Phylogenet. Evol. 2008, 49, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Q.Y.; Soltis, D.E.; Soltis, P.S. The eastern Asian and eastern and western North American floristic disjunction: Congruent phylogenetic patterns in seven diverse genera. Mol. Phylogenet. Evol. 1998, 10, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Wen, J. Evolution of eastern Asian and eastern North American disjunct distributions in flowering plants. Annu. Rev. Ecol. Syst. 1999, 30, 421–455. [Google Scholar] [CrossRef]
- Wolfe, J.A. Some aspects of plant geography in the Northern Hemisphere during the late Cretaceous and Tertiary. Ann. Mo. Bot. Gard. 1975, 62, 264–279. [Google Scholar] [CrossRef]
- Tiffney, B.H.; Manchester, S.R. The use of geological and paleontological evidence in evaluating plant phylogeographic hypotheses in the Northern Hemisphere Tertiary. Int. J. Plant Sci. 2001, 162, 48–52. [Google Scholar] [CrossRef]
- Li, H.L. Floristic relationships between eastern Asia and eastern North America. Trans. Am. Philos. Soc. 1952, 42, 371–429. [Google Scholar] [CrossRef]
- Fukuoka, N.; Kurosaki, N. Phytogeographical notes on some species of west Honshu, Japan 5. Shoei Jr. Col. Ann. Rep. Stud. 1985, 17, 61–71. [Google Scholar]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Boil. 2013, 13, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Ruhsam, M.; Rai, H.S.; Mathews, S.; Ross, T.G.; Graham, S.W.; Raubeson, L.A.; Mei, W.; Thomas, P.I.; Gardner, M.F.; Ennos, R.A. Does complete plastid genome sequencing improve species discrimination and phylogenetic resolution in Araucaria? Mol. Ecol. Res. 2015, 15, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Firetti, F.; Zuntini, A.R.; Gaiarsa, J.W.; Oliveira, R.S.; Lohmann, L.G.; Van Sluys, M.A. Complete chloroplast genome sequences contribute to plant species delimitation: A case study of the Anemopaegma species complex. Am. J. Bot. 2017, 104, 1493–1509. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; Claude, W.; Leebensmack, J.; Guisingerbellian, M.; Haberle, R.C.; Hansen, A.; Chumley, T.W. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Ma, P.; Li, H.; Li, D. Complete plastid genome sequencing of four Tilia species (Malvaceae): A comparative analysis and phylogenetic implications. PLoS ONE 2015, 10, e0142705. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.B.; Alverson, A.J.; Wu, M.; Palmer, J.D.; Taylor, D.R. Recent acceleration of plastid sequence and structural evolution coincides with extreme mitochondrial divergence in the angiosperm genus Silene. Genome Biol. Evol. 2012, 4, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qu, X.; Chen, S.; Li, D.; Yi, T. Plastomes of Mimosoideae: Structural and size variation, sequence divergence, and phylogenetic implication. Tree Genet. Genom. 2017, 13, 41–56. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.E.; Li, C. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 3, e57607. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Li, H.; Milne, R.I.; Zhang, T.; Ma, P.; Yang, J.; Li, D.; Gao, L. Comparative analyses of plastid genomes from fourteen Cornales species: Inferences for phylogenetic relationships and genome evolution. BMC Genom. 2017, 18, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.R.; Dastidar, S.G.; Cai, Z.; Penaflor, C.; Kuehl, J.V.; Boore, J.L.; Jansen, R.K. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 2007, 45, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Huo, N.; Dong, L.; Wang, Y.; Zhang, S.; Young, H.A.; Feng, X.; Gu, Y.Q. Complete chloroplast genome sequences of mongolia medicine Artemisia frigida and phylogenetic relationships with other plants. PLoS ONE 2013, 8, e57533. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.F.; Zanis, M.J.; Emery, N.C. Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Am. J. Bot. 2014, 101, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, X.; Liu, G.; Yin, Y.; Chen, K.; Yun, Q.; Zhao, D.G.; Almssallem, I.S.; Yu, J. The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS ONE 2010, 5, e12762. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cui, L.; Feng, K.; Deng, P.; Du, X.; Wan, F.; Weining, S.; Nie, X. Comparative analysis of asteraceae chloroplast genomes: Structural organization, RNA editing and evolution. Plant Mol. Biol. Rep. 2015, 33, 1526–1538. [Google Scholar] [CrossRef]
- Palmer, J.D. Chloroplast DNA exists in two orientations. Nature 1983, 301, 92–93. [Google Scholar] [CrossRef]
- Wolfe, A.D.; Randle, C.P. Recombination, heteroplasmy, haplotype polymorphism, and paralogy in plastid genes: Implications for plant molecular systematics. Syst Bot. 2004, 29, 1011–1020. [Google Scholar] [CrossRef]
- Walker, J.F.; Jansen, R.K.; Zanis, M.J.; Emery, N.C. Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Am. J. Bot. 2015, 102, 1751–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Downie, S.R.; Jansen, R.K. A comparative analysis of whole plastid genomes from the apiales: Expansion and contraction of the inverted repeat, mitochondrial to plastid transfer of DNA, and identification of highly divergent noncoding regions. Sys. Bot. 2015, 40, 336–351. [Google Scholar] [CrossRef]
- Wang, R.J.; Cheng, C.L.; Chang, C.C.; Wu, C.L.; Su, T.M.; Chaw, S. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Nazareno, A.G.; Carlsen, M.; Lohmann, L.G. Complete chloroplast genome of tanaecium tetragonolobum: The first Bignoniaceae plastome. PLoS ONE 2015, 10, e0129930. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tang, P.; Li, Z.; Li, D.; Liu, Y.; Huang, H. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar] [CrossRef] [PubMed]
- Khakhlova, O.; Bock, R. Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 2006, 46, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ma, P.; Li, D. High-throughput sequencing of six bamboo chloroplast genomes: Phylogenetic implications for temperate woody bamboos (Poaceae: Bambusoideae). PLoS ONE 2011, 6, e20596. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.S.; Chung, M.G.; Park, S. The Complete Chloroplast Genome Sequences of Three Veroniceae Species (Plantaginaceae): Comparative Analysis and Highly Divergent Regions. Front. Plant Sci. 2016, 7, 355–394. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.S.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lu, R.; Xu, W.; Ohitoma, T.; Cai, M.; Qiu, Y.; Cameron, K.M.; Fu, C. Comparative genomics and phylogenomics of east Asian tulips (amana, Liliaceae). Front. Plant Sci. 2017, 8, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.E.; Linder, H.P.; Donoghue, M.J. The historical biogeography of Scabiosa (Dipsacaceae): Implications for Old World plant disjunctions. J. Biol. 2012, 39, 1086–1100. [Google Scholar] [CrossRef]
- Yue, J.; Sun, H.; Baum, D.A.; Jianhua, L.I.; Alshehbaz, I.A.; Ree, R.H. Molecular phylogeny of Solms-laubachia (Brassicaceae) s.l., based on multiple nuclear and plastid DNA sequences, and its biogeographic implications. J. Syst. Evol. 2009, 47, 402–415. [Google Scholar] [CrossRef]
- Kurtz, S.; Schleiermacher, C. REPuter: Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Shi, C.; Liu, Y.; Mao, S.Y.; Gao, L.Z. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships. BMC Evol. Biol. 2014, 14, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yin, J.; Guo, H.; Zhang, Y.; Xiao, W.; Sun, C.; Wu, J.; Qu, X.; Yu, J.; Wang, X. The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Front. Plant Sci. 2015, 5, 696–698. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.C.; Lee, J.; Yu, Y.; Yang, K.; Choi, B.S.; Koh, H.J.; Waminal, N.E.; Choi, H.I.; Kim, N.H. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 2015, 5, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.I.; Abbott, R.J. The origin and evolution of tertiary relict floras. Adv. Bot. Res 2002, 38, 281–314. [Google Scholar]
- Tiffney, B.H. Perspectives on the origin of the floristic similarity between eastern Asia and eastern North America. J. Arn. Arb. 1985, 66, 73–94. [Google Scholar] [CrossRef]
- Xiang, Q.; Soltis, D.E.; Soltis, P.S.; Manchester, S.R.; Crawford, D.J. Timing the eastern Asian-eastern North American floristic disjunction: Molecular clock corroborates paleontological estimates. Mol. Phylogenet. Evol. 2000, 15, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Birky, C.W. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef] [PubMed]
- Cronn, R.; Liston, A.; Parks, M.; Gernandt, D.S.; Shen, R.; Mockler, T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008, 36, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Bio. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. Gatew. Comput. Environ. Workshop 2010, 3, 1–8. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmann, N.; Wolf, E.M.; Lysak, M.A.; Koch, M.A. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell 2015, 27, 2770–2784. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Hu, Q.; Alshehbaz, I.A.; Luo, X.; Zeng, T.; Guo, X.; Liu, J. Species delimitation and interspecific relationships of the genus Orychophragmus (Brassicaceae) inferred from whole chloroplast genomes. Front. Plant Sci. 2016, 7, 1826–2884. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J. Tracer v.1.4. Encycl. Atmos. Sci. 2007, 141, 2297–2305. [Google Scholar]
- Bouckaert, R.R.; Heled, J.; Kuhnert, D.; Vaughan, T.G.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for bayesian evolutionary analysis. PLoS Computat. Biol. 2014, 10, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Harris, A.J.; Blair, C.; He, X. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography. Mol. Phylogenet. Evol. 2015, 87, 46–49. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category of Genes | Groups of Genes | Names of Genes |
|---|---|---|
| Self-replication | rRNA genes | rrn16(×2), rrn23(×2), rrn4.5(×2), rrn 5(×2) |
| tRNA genes | trnA-UGC *(×2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCC *, trnH-GUG(×2), trnI-CAU(×2), trnI-GAU *(×2), trnK-UUU *, trnL-CAA(×2), trnL-UAA *, trnL-UAG, trnM-CAU, trnN-GUU(×2), trnP-UGG, trnQ-UUG, trnR-ACG(×2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC(×2), trnV-UAC *, trnW-CCA, trnY-FUA | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7(×2), rps8, rps11, rps12 **(×2), rps14, rps15, rps16 *, rps18, rps19(×2) | |
| Large subunit of ribosome | rpl2 *(×2), rpl14, rpl16 *, rpl20, rpl22(×2), rpl23(×2), rpl32, rpl33, rpl36 | |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1 *, rpoC2 | |
| Genes for photosynthesis | Subunit of NADH-dehydrogenase | ndhA *, ndhB *(×2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhI, ndhH, ndhJ, hdhK |
| Subunit of Photosystem 1 | psaA, psaB, psaC, psaI, psaJ, ycf3 ** | |
| Subunit of Photosystem 2 | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT | |
| Subunits of cytochrome b/f complex | petA, petB *, petD *, petG, petL, petN | |
| Subunits of ATP synthase | atpA, atpB, atpE, atpF *, atpH, atpI | |
| Large subunit of rubisco | rbcL | |
| Other genes | Maturase | matK, |
| Protease | clpP ** | |
| Envelope membrane protein | cemA | |
| Subunit of Acetyl-CoA-carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Translation initiation factor IF-1 | infA | |
| Genes of unknown function | Open reading frames (ORF, ycf) | ycf1, ycf2(×2), ycf4, lhbA |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Q.; Ye, W.; Lu, R.; Xu, W.; Qiu, Y. Phylogenomic and Comparative Analyses of Complete Plastomes of Croomia and Stemona (Stemonaceae). Int. J. Mol. Sci. 2018, 19, 2383. https://doi.org/10.3390/ijms19082383
Lu Q, Ye W, Lu R, Xu W, Qiu Y. Phylogenomic and Comparative Analyses of Complete Plastomes of Croomia and Stemona (Stemonaceae). International Journal of Molecular Sciences. 2018; 19(8):2383. https://doi.org/10.3390/ijms19082383
Chicago/Turabian StyleLu, Qixiang, Wenqing Ye, Ruisen Lu, Wuqin Xu, and Yingxiong Qiu. 2018. "Phylogenomic and Comparative Analyses of Complete Plastomes of Croomia and Stemona (Stemonaceae)" International Journal of Molecular Sciences 19, no. 8: 2383. https://doi.org/10.3390/ijms19082383
APA StyleLu, Q., Ye, W., Lu, R., Xu, W., & Qiu, Y. (2018). Phylogenomic and Comparative Analyses of Complete Plastomes of Croomia and Stemona (Stemonaceae). International Journal of Molecular Sciences, 19(8), 2383. https://doi.org/10.3390/ijms19082383