1. Introduction
Flavonoids are naturally occurring compounds that contain a number of phenolic hydroxyl groups attached to ring structures designated as A, B, and C (
Figure 1A). The structure of flavonoids is usually characterized by two aromatic rings (A and B) joined by a three-carbon linked C-pyrone ring (C) to form a C6–C3–C6 skeletal unit (
Figure 1A). The bioactivity of flavonoids has attracted academic interest because of its potential health benefits for humans. Diets rich in fruits and vegetables exert protective effects against cardiovascular diseases (CVDs) and certain forms of cancer [
1]. Flavonoids have been used as valuable therapeutic agents in modern and traditional medicine.
Morin is a well-known bioactive constituent belonging to the flavonol group and is found in old fustic (
Chlorophora tinctoria), Osage orange (
Maclura pomifera), almond, mill (
Prunus dulcis), fig (
Chlorophora tinctoria), onion, apple, and other Moraceae, which are used as dietary agents and herbal medicines [
2,
3]. Morin exhibits various biological activities such as antimutagenesis, anti-inflammation, antiarthritis, and cardioprotective activities, and inhibits xanthine oxidase activity and cell proliferation [
4,
5,
6]. Morin has attracted considerable interest because of its antitumor activity in vitro and in vivo [
7].
Dietary factors play key roles in the development and prevention of various human diseases, such as myocardial infarction and ischemic stroke. The risk of CVDs have been reduced with intake of dietary flavonoids [
8]. Platelet adherence and aggregation is believed to be initiated intraluminal thrombosis and thus, these events may play a crucial role in atherothrombotic processes in addition to mediating hemostasis [
9]. Moreover, platelets are the first-line defense against hemorrhage and important for maintaining the integrity of the vascular system. Upon platelet activation, several mediators such as adenosine triphosphate (ATP) and thromboxane A
2 are released in conjunction with intracellular Ca
2+ ([Ca
2+]i) mobilization. These processes further attract other platelets toward the injured endothelium and therefore cause the thickening of the initial platelet monolayer. Finally, fibrinogen binds to its specific platelet receptor of integrin α
IIbβ
3, thereby completing the final common pathway for platelet aggregation.
Tzeng et al. [
10] demonstrated that morin hydrate exhibited potent bioactivity in the inhibition of rabbit platelet aggregation stimulated by arachidonic acid (AA). In our preliminary study, we observed that 20 µM of morin hydrate significantly inhibited platelet aggregation in washed human platelets. However, the effect of morin hydrate on platelet activation remained to be thoroughly investigated. Therefore, in the current study, we systematically examined in vitro and in vivo effects of morin hydrate on washed human platelets and experimental mice to further characterize the detailed mechanisms of morin hydrate–mediated inhibition of platelet activation.
3. Discussion
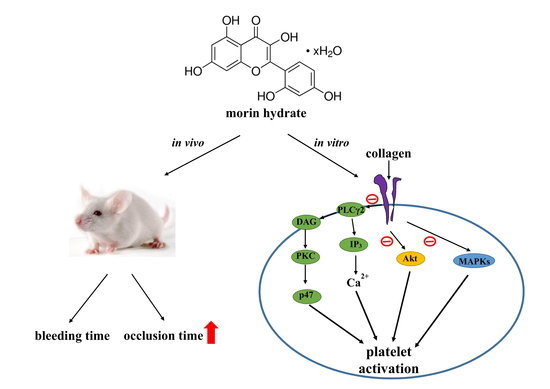
Our study was the first to demonstrate that morin hydrate shows in vitro antiplatelet effect and effectively inhibit arterial thrombosis in vivo. In general, dietary intake from the natural sources is likely insufficient for attaining plasma concentrations that can inhibit platelet activation in vivo, however, several nontoxic prophylactic agents such as food products and nutritional supplements is ideal for stopping atherothrombotic events. Thus, morin hydrate may signify a novel antithrombotic agent for use in humans.
In the present study, morin hydrate effectively inhibited platelet aggregation stimulated by collagen but not that stimulated by thrombin, AA, or U46619, indicating that this compound inhibits platelet aggregation through a markedly PLC-dependent mechanism. Various physiological stimuli (e.g., thrombin, collagen, ADP, and epinephrine) activate platelets, and these stimulators are believed to exert their effects via interaction with specific receptors on platelet membranes. The matrix protein collagen is present in the vascular subendothelium and vessel wall and acts as a substrate for platelet adhesion; it is also an endogenous platelet activator. Among the platelet receptors known to interact directly with collagen, integrin α
2β
1 and glycoprotein (GP) VI [
15] appear to play a key role and have recently gained academic attention. GP VI is widely recognized as a requisite factor for formation of platelet aggregates on collagen surfaces under blood flow [
16]. Integrin α
2β
1 is another major collagen receptor on endothelial cells and platelets. In cells expressing integrin α
2β
1, many signals (including tyrosine phosphorylation and matrix remodeling) are activated after cell adhesion to collagen [
17]. Recent findings have suggested that integrin α
2β
1 and GP VI might contribute to the overall processes of platelet adhesion and activation [
18].
Activation of platelets by collagen substantially alters phospholipase activation. PLC activation is an early event in response to numerous extracellular stimuli. Upon activation, PLC produces two crucial second messengers, namely DAG and inositol trisphosphate (IP
3), both of which play key roles in many signaling pathways, including activation of protein kinase C (PKC) and protein kinase D (PKD) and induction of calcium influx [
19]. The signaling axis of PLC/PKC/PKD was shown to play a key role in many signaling pathways [
20]. In response to various extracellular stimuli, there are 13 phosphatidylinositide-specific PLCs that are divided into six subgroups: PLCβ, PLCγ, PLCδ, PLCε, PLCξ, and PLCη [
21]. The PLCγ family comprises isozymes 1 and 2. PLCγ2 is involved in collagen-dependent signaling on platelets [
21]. In our study, morin hydrate diminished the activation of the PLCγ2–PKC cascade stimulated by collagen, suggesting that the morin hydrate–mediated inhibition of platelet activation involves PLCγ2 downstream signaling; moreover, this result explains why morin hydrate was more efficacious in inhibiting collagen induced platelet aggregation than that induced by thrombin, U46619, or AA.
Platelets adhere to the subendothelial matrix protein collagen, which alters their shape and releases granular contents of ATP, Ca
2+, and P-selectin. P-selectin is an adhesion molecule kept in the platelets α-granules, upon activation it is expressed on a platelet surface membrane and successively expressed on the external membrane through membrane flipping. P-selectin mediates the initial formation of platelet aggregates and simplifies the development of large platelet aggregates [
22]. Agonists such as collagen, thrombin, and AA activate [Ca
2+]i to phosphorylate the Ca
2+/calmodulin-dependent myosin light chain (20 kDa), which is involved in the secretion of granule contents such as serotonin and ATP [
23], as well as platelet aggregation. So, suppression of [Ca
2+]i mobilization and ATP production is critical for assessing the antiplatelet effects of a compound.
Specific MAPK kinases (MEKs), specifically, MEK1/2, MEK3/6, and MEK4/7 activate MAPKs, such as ERKs, p38 MAPK, and JNKs, respectively [
24]. ERK2 activation is involved in platelet aggregation needing prior ATP release, which triggers P
2X
1-mediated Ca
2+ influx and activates ERK2, thereby increasing the phosphorylation of myosin light chain kinase [
25]. JNK1 is recently identified in platelets and it activated by several agonists, including collagen, thrombin, and ADP [
26], however its role in platelets is poorly recognized. Furthermore, a reduced integrin α
IIbβ
3 activation and severe granule secretion impairment was proved in JNK
−/− platelets [
25]. Additionally, cytosolic phospholipase A
2 (cPLA
2), a substrate of p38 MAPK activity is induced by agonists such as von Willebrand factor and thrombin [
25]. Therefore, p38 MAPK is consider important for cPLA
2 stimulation [
27]. These literatures revealed reasonable explanation is why morin hydrate inhibited ERK2 and JNK1 activation largely than p38 MAPK activation.
Akt is a downstream effector of phosphoinositide 3 (PI3)-kinase. A defective agonist-induced platelet activation was found in Akt-knockout mice, proposing that Akt regulates platelet activation and such regulation may have consequences related to thrombosis [
13]. Among the three mammalian Akt isoforms of Akt 1, 2, and 3, the first two isoforms were detected in human platelets [
28]. Akt inhibitors have usually reported similar roles of Akt 1 and 2 in human platelet activation. Therefore, protein kinases for Akt activation, particularly PI3-kinase β, may be appropriate targets for the development of antithrombotic therapies. Our previous study found PI3-kinase/Akt and MAPKs are mutually activated as upstream regulators of PKC in activated platelets [
29].
Reactive oxygen species (ROS) produced through platelet activation (i.e., H
2O
2 and OH
·) might affect cells that they come into contact with such as endothelial cells, thereby enhancing platelet reactivity during thrombus formation. Free radicals act as secondary signals that increase [Ca
2+]i levels during the initial phase of platelet activation, and PKC is involved in receptor-mediated production of free radicals in platelets [
30]. In addition, H
2O
2 produced by platelets is converted into OH
· because platelet aggregation is inhibited by OH
· scavengers [
30]. Morin was reported to scavenge ROS and free radicals of oxygen in an indirect analysis study [
31]. Our ESR spectrometry results provided direct evidence that morin hydrate significantly reduced OH
· formation in platelet suspensions but had no effect in a cell-free system.
In this study on thrombosis, mesenteric venules were continuously irradiated by fluorescein sodium throughout the experimental period, leading to strong damage to the endothelial cells. Here, 10 mg/kg of morin hydrate significantly prolonged the occlusion times; this effect may be mediated, at least partially, by inhibition of platelet activation. In addition, we used the tail transection mouse model to examine the effects of morin hydrate on bleeding time in vivo. Although aspirin is the most effective antiplatelet drug prescribed for preventing or treating cardiovascular and cerebrovascular diseases, it causes unwanted prolongation of bleeding time. In this model, the bleeding time of the morin hydrate–treated mice was not significantly different to the solvent control, indicating that morin hydrate possesses antiplatelet activity in vivo but had no effect on bleeding time. Liu et al. [
32] reported that treatment with 20 mg/kg of morin hydrate markedly reduced myocardial ischemia-reperfusion injury (MIRI) in rats. Although results in animal species and experimental models have differed, morin hydrate evidently possesses a more potent ability to inhibit arterial thrombosis than protect against MIRI.
The findings of the present study revealed that morin hydrate plays a novel role in inhibiting platelet activation in vitro and in vivo, suggesting that it can be used in prophylactic applications. Generally, a nutritional or dietary supplement is required to produce a prophylactic effect in humans. However, selection of doses for time-course treatments may be confounded by variation in responses among users. This study provided new insights into the role of morin hydrate in blocking collagen-specific signaling events involved in platelet activation. However, the involvement of other mechanisms yet to be identified in morin hydrate–mediated inhibition of platelet activation requires investigation.
4. Materials and Methods
4.1. Chemicals and Reagents
Morin hydrate, thrombin, collagen, AA, luciferin–luciferase, U46619, heparin, prostaglandin E1 (PGE1), 5,5-dimethyl-1-pyrroline N-oxide (DMPO), and bovine serum albumin (BSA) were purchased from Sigma (St. Louis, MO, USA). Fura-2AM was obtained from Molecular Probes (Eugene, OR, USA). An anti-phospho-p38 mitogen-activated protein kinase (MAPK) Ser182 monoclonal antibody (mAb) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-p38 MAPK, anti-phospho-c-Jun N-terminal kinase (JNK) (Thr183/Tyr185), and anti-p44/42 extracellular signal-regulated kinase (ERK) mAbs, as well as anti-phospholipase Cγ2 (PLCγ2), anti-phospho (Tyr759) PLCγ2, anti-phospho-(Ser) protein kinase C (PKC) substrate (pleckstrin; p-p47), anti-JNK, and anti-phospho-p44/p42 ERK (Thr202/Tyr204) polyclonal antibodies (pAbs) were purchased from Cell Signaling (Beverly, MA, USA). Anti-phospho-protein kinase B (Akt) (Ser473) and anti-Akt mAbs were purchased from Biovision (Mountain View, CA, USA). An anti-pleckstrin (p47) pAb was purchased from GeneTex (Irvine, CA, USA). A Hybond-P polyvinylidene fluoride (PVDF) membrane, an enhanced chemiluminescence Western blotting detection reagent, horseradish peroxidase (HRP)-conjugated donkey anti-rabbit immunoglobulin G (IgG), and sheep anti-mouse IgG were purchased from Amersham (Buckinghamshire, UK). A fluorescein isothiocyanate (FITC) anti-human CD42P (P-selectin) mAb was obtained from BioLegend (San Diego, CA, USA). Morin hydrate was dissolved in 0.1% dimethyl sulfoxide (DMSO) and stored at 4 °C.
4.2. Platelet Aggregation
This study was approved by the Institutional Review Board of Taipei Medical University (N201612050; 20/January/2017) and conformed to the directives of the Declaration of Helsinki. All human volunteers involved in this study provided informed consent. Human platelet suspensions were prepared as described previously [
9]. Human blood samples were obtained from adult volunteers who refrained from use of drugs or other substances that could have interfered with the experiment for at least 14 days before sample collection; the collected blood samples were mixed with an acid-citrate-dextrose solution. After centrifugation, platelet-rich plasma (PRP) was supplemented with 0.5 μM of PGE
1 and 6.4 IU/mL of heparin. Tyrode’s solution containing 3.5 mg/mL of BSA was used to prepare the final suspension of washed human platelets. The final Ca
2+ concentration in the Tyrode’s solution was 1 mM. A platelet aggregation study was conducted using a lumi-aggregometer (Payton Associates, Scarborough, ON, Canada), as described previously [
9]. An isovolumetric solvent control (0.1% DMSO) or morin hydrate was preincubated with platelet suspensions (3.6 × 10
8 cells/mL) for 3 min before the addition of the agonists (i.e., collagen). The extent of platelet aggregation was calculated as the percentage compared with individual control (without morin hydrate) expressed in light transmission units after the reaction had proceeded for 6 min. For an ATP release assay, 20 μL of luciferin–luciferase was added 1 min before the addition of collagen (1 µg/mL), and the amount of ATP released was compared with that released by the control (without morin hydrate).
4.3. Measurement of Relative [Ca2+]i Mobilization
The relative [Ca
2+]i concentration was determined using Fura-2AM, as described previously [
9]. Concisely, citrated whole blood was centrifuged at 120×
g for 10 min and the PRP was collected and incubated with Fura-2AM (5 μM) for 1 h. The Fura-2AM-loaded platelets were preincubated with morin hydrate (40 and 80 µM) in the presence of 1 mM of CaCl
2 and then stimulated with collagen (1 µg/mL). The Fura-2 fluorescence was measured using a spectrofluorometer (Hitachi FL Spectrophotometer F-4500, Tokyo, Japan) at excitation wavelengths of 340 and 380 nm and an emission wavelength of 510 nm.
4.4. Flow Cytometric Analysis of Surface P-selectin Expression
Washed platelets were prepared as described in the preceding subsection and aliquots of platelet suspensions (3.6 × 108 cells/mL) were preincubated with the solvent control (0.1% DMSO) or morin hydrate (40 and 80 µM) and FITC-P-selectin (2 µg/mL) for 3 min; collagen (1 µg/mL) was added to trigger platelet activation. The suspensions were then assayed for fluorescein-labeled platelets by using a flow cytometer (FACScan System, Becton Dickinson, San Jose, CA, USA). Data were collected from 50,000 platelets per experimental group. All experiments were repeated at least four times to ensure reproducibility.
4.5. Detection of Lactate Dehydrogenase
Washed platelets (3.6 × 108 cells/mL) were preincubated with the solvent control (0.1% DMSO) or morin hydrate (40, 80, 100 μM) for 20 min at 37 °C. An aliquot of the supernatant (10 µL) was deposited on a Fuji Dri-Chem slide LDH-PIII (Fuji, Tokyo, Japan) and the absorbance wavelength was read at 540 nm by using an ultraviolet–visible spectrophotometer (UV-160; Shimadzu, Japan). A maximal value of lactate dehydrogenase (LDH) was recorded in the sonicated platelets (Max).
4.6. Immunoblotting of Protein Phosphorylation
Washed platelets (1.2 × 109 cells/mL) were preincubated with the solvent control (0.1% DMSO) or morin hydrate (40 and 80 µM) for 3 min. Subsequently, collagen (1 µg/mL) was added to trigger platelet activation. The reaction was then stopped and the platelets were immediately resuspended in 200 μL of lysis buffer. Samples containing 80 μg of protein were separated through 12% sodium dodecyl sulfate gel electrophoresis, and the proteins were electrotransferred to PVDF membranes by using a Bio-Rad semidry transfer unit (Bio-Rad, Hercules, CA, USA). The blots were then blocked through treatment with Tris-buffered saline in Tween 20 (TBST; 10 mM of Tris-base, 100 mM of NaCl, and 0.01% Tween 20) containing 5% BSA for 1 h and were probed with various primary antibodies. The membranes were incubated with HRP-conjugated anti-mouse IgG or anti-rabbit IgG (diluted 1:3000 in TBST) for 1 h. An enhanced chemiluminescence system was used to detect immunoreactive bands, whose optical density was quantified using Bio-profil Biolight (version V2000.01; Vilber Lourmat, Marne-la-Vallée, France).
4.7. Measurement of OH· Formation in the Platelet Suspensions or Fenton Reaction Solution Through Electron Spin Resonance Spectrometry
Electron spin resonance (ESR) spectrometry was performed using a Bruker EMX ESR spectrometer (Bruker, Billerica, MA, USA), as described previously [
33]. Suspensions of washed platelets (3.6 × 10
8 cells/mL) or the Fenton reagent (50 μM FeSO
4 + 2 mM H
2O
2) were preincubated with 0.1% DMSO or morin hydrate (40 and 80 µM) for 3 min. Subsequently, collagen (1 μg/mL) was added into platelet suspensions to trigger platelet activation for 5 min. Before ESR spectrometry, 100 μM of DMPO was added to both solutions. ESR spectra were recorded using a quartz flat cell designed for aqueous solutions. The spectrometer was operated under the following conditions: power, 20 mW; frequency, 9.78 GHz; scan range, 100 G; and receiver gain, 5 × 10
4. The modulation amplitude was 1G, the time constant was 164 ms, and scanning was performed for 42 s; each ESR spectrum obtained was the sum of four scans.
4.8. Measurement of Sodium Fluorescein-Induced Thrombus Formation in Mouse Mesenteric Microvessels
Male ICR mice (aged 6 weeks) were anesthetized using a mixture containing 75% air and 3% isoflurane maintained in 25% oxygen; the mice’s external jugular veins were then cannulated with a PE-10 tube for administration of dye and drugs intravenously [
34]. Venules (30–40 µm) were irradiated at wavelengths of <520 nm to produce a microthrombus. Two morin hydrate doses (5 and 10 mg/kg) were administered 1 min following sodium fluorescein (15 µg/kg) administration, and the time required for the thrombus to occlude the microvessel (occlusion time) was recorded. In this experiment, the method applied to the thrombogenic animal model conformed to the Guide for the Care and Use of Laboratory Animals (8th edition, 2011), and we received an affidavit of approval for the animal use protocol from Taipei Medical University (LAC-2016-0276; 01/August/2017).
4.9. Measurement of Bleeding Time in Mouse Tail Vein
Bleeding time was measured through transection of the tails of the male ICR mice. In brief, after 30 min of administration of 5 or 10 mg/kg of morin hydrate intraperitoneally, the tails of the mice were cut 3 mm from the tip. The tails were then immediately placed into a tube filled with normal saline at 37 °C to measure the bleeding time, which was recorded until the bleeding completely stopped. In the animal experiments, the method applied to the animal model conformed to the Guide for the Care and Use of Laboratory Animals (8th edition, 2011), and we received an affidavit of approval for the animal use protocol from Taipei Medical University (LAC-2016-0276; 01/August/2017).
4.10. Statistical Analysis
The experimental results are expressed as means ± standard errors of means alongside the number of observations (n); n refers to the number of experiments; each experiment was performed using different blood donors. The unpaired Student’s t test was used to determine the significance of differences between control and experimental mice. Differences between groups in other experiments were assessed using an analysis of variance (ANOVA). When the ANOVA results indicated significant differences among group means, the groups were compared using the Student–Newman–Keuls method. A p value of <0.05 indicated statistical significance. Statistical analyses were performed using SAS (version 9.2; SAS Inc., Cary, NC, USA).

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





