Implications of Metal Binding and Asparagine Deamidation for Amyloid Formation


Abstract
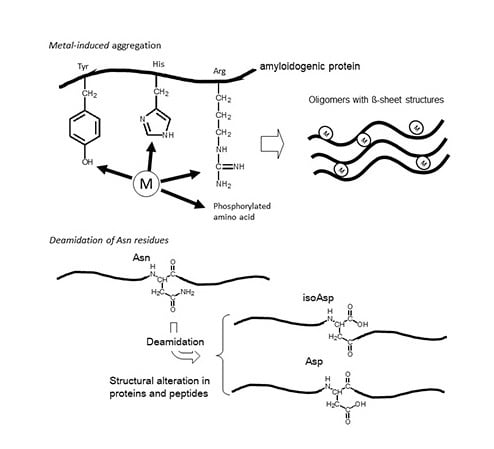
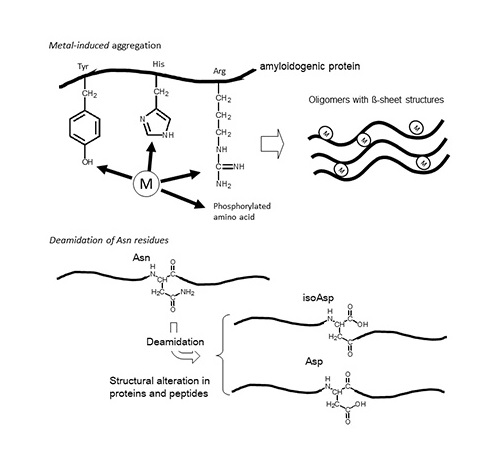
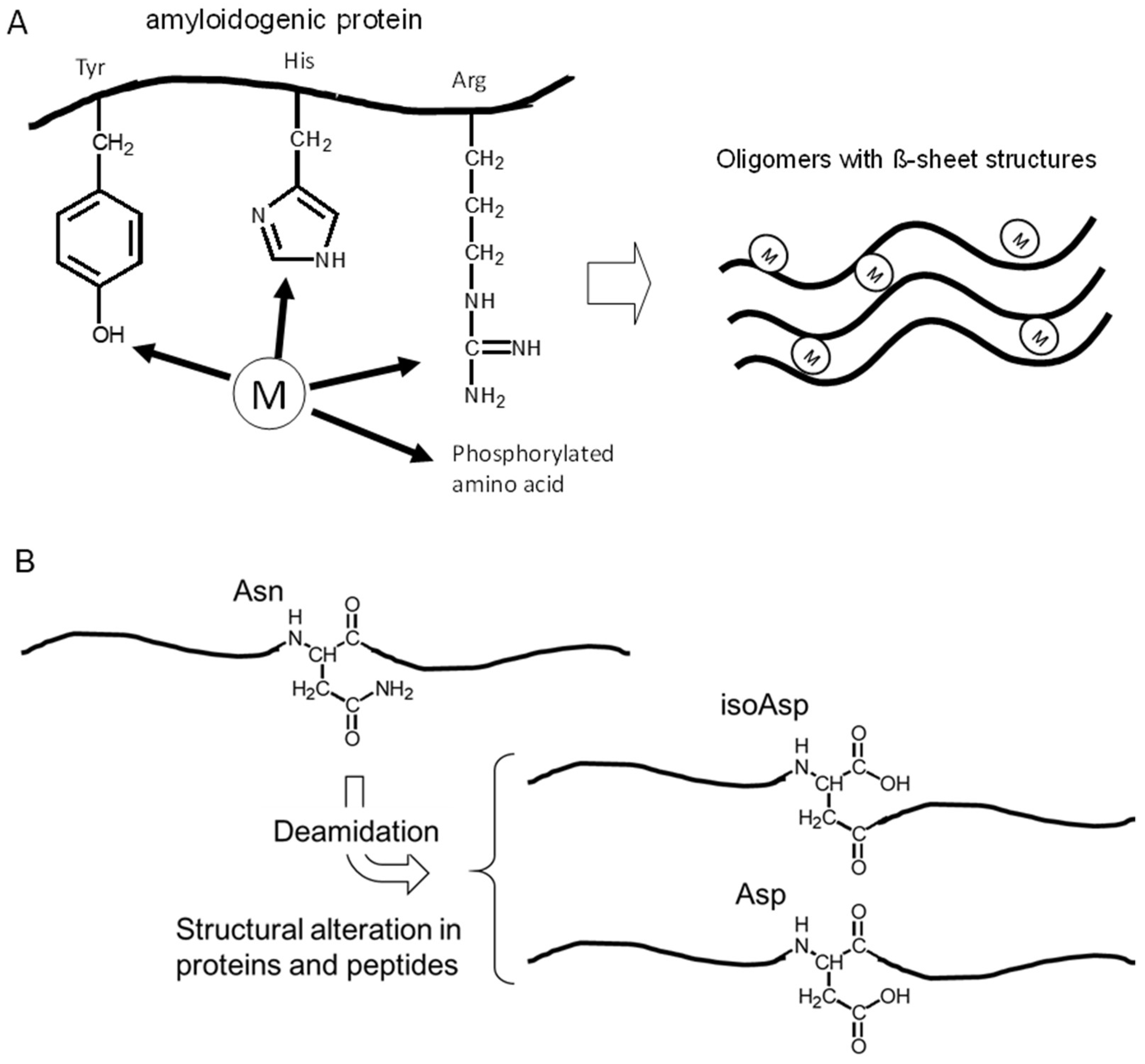
:
1. Introduction
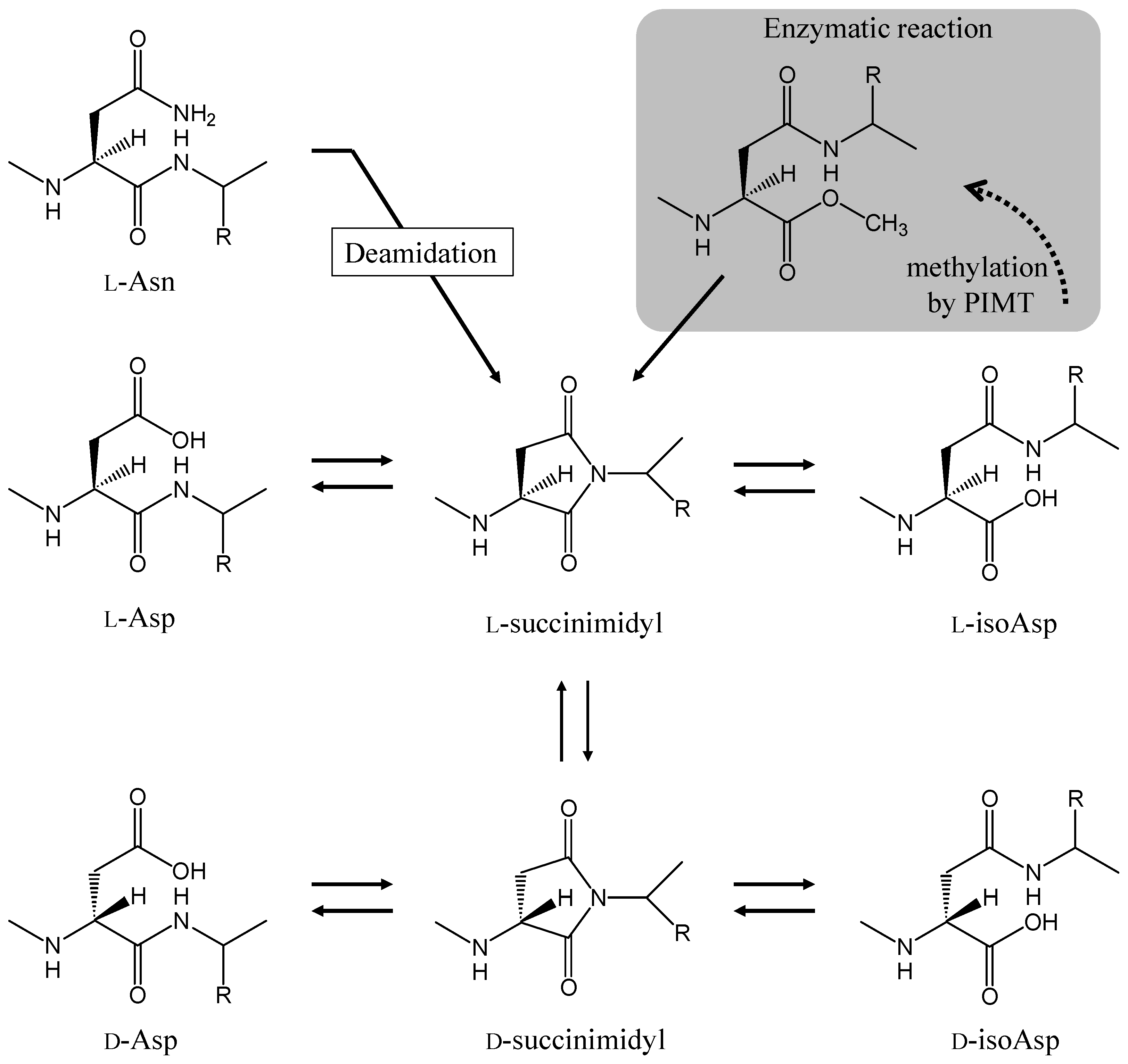
2. Asparagine Deamidation and its Biological Significance
3. Alzheimer’s Disease
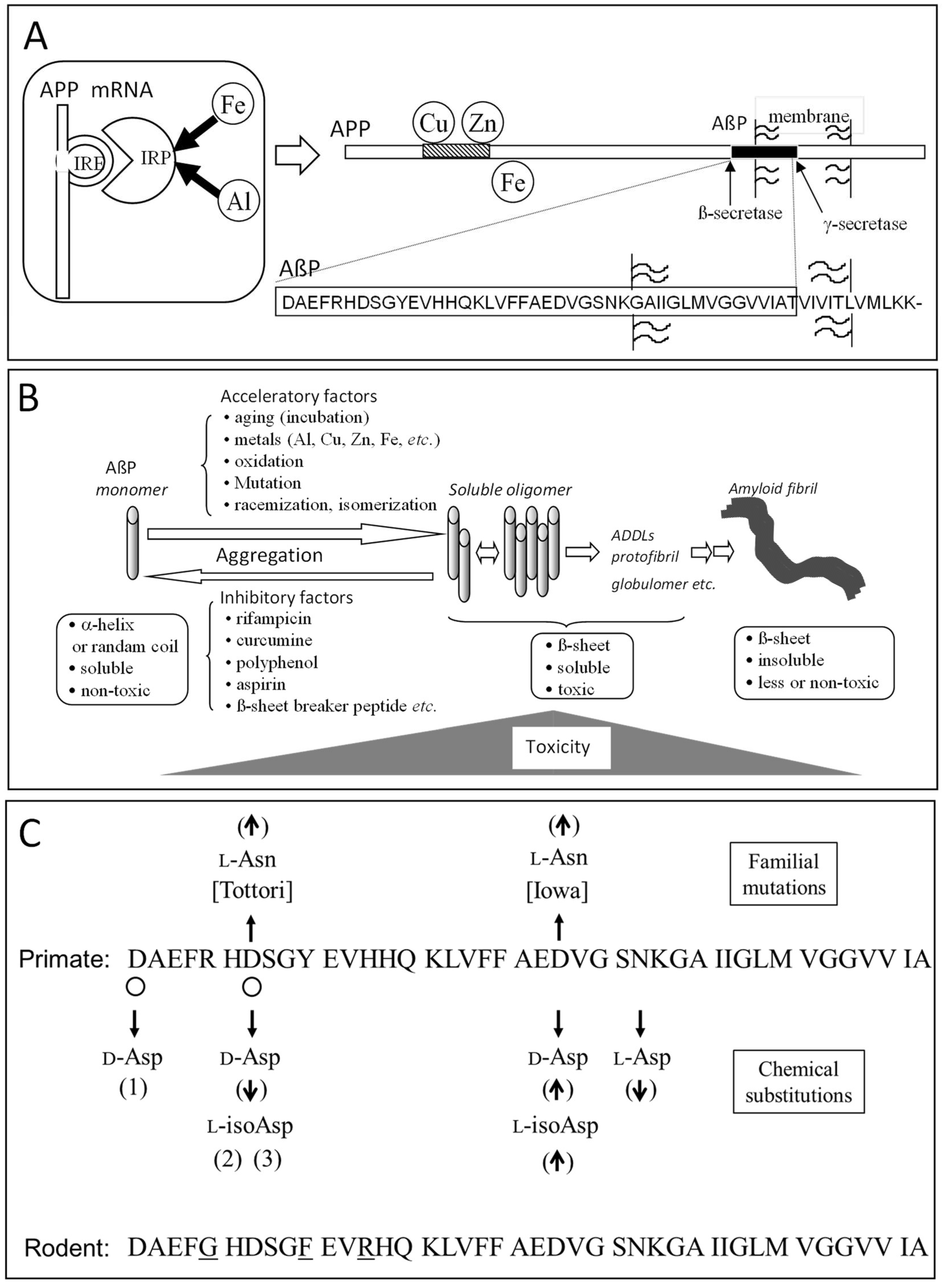
3.1. Alzheimer’s Disease and β-Amyloid Protein
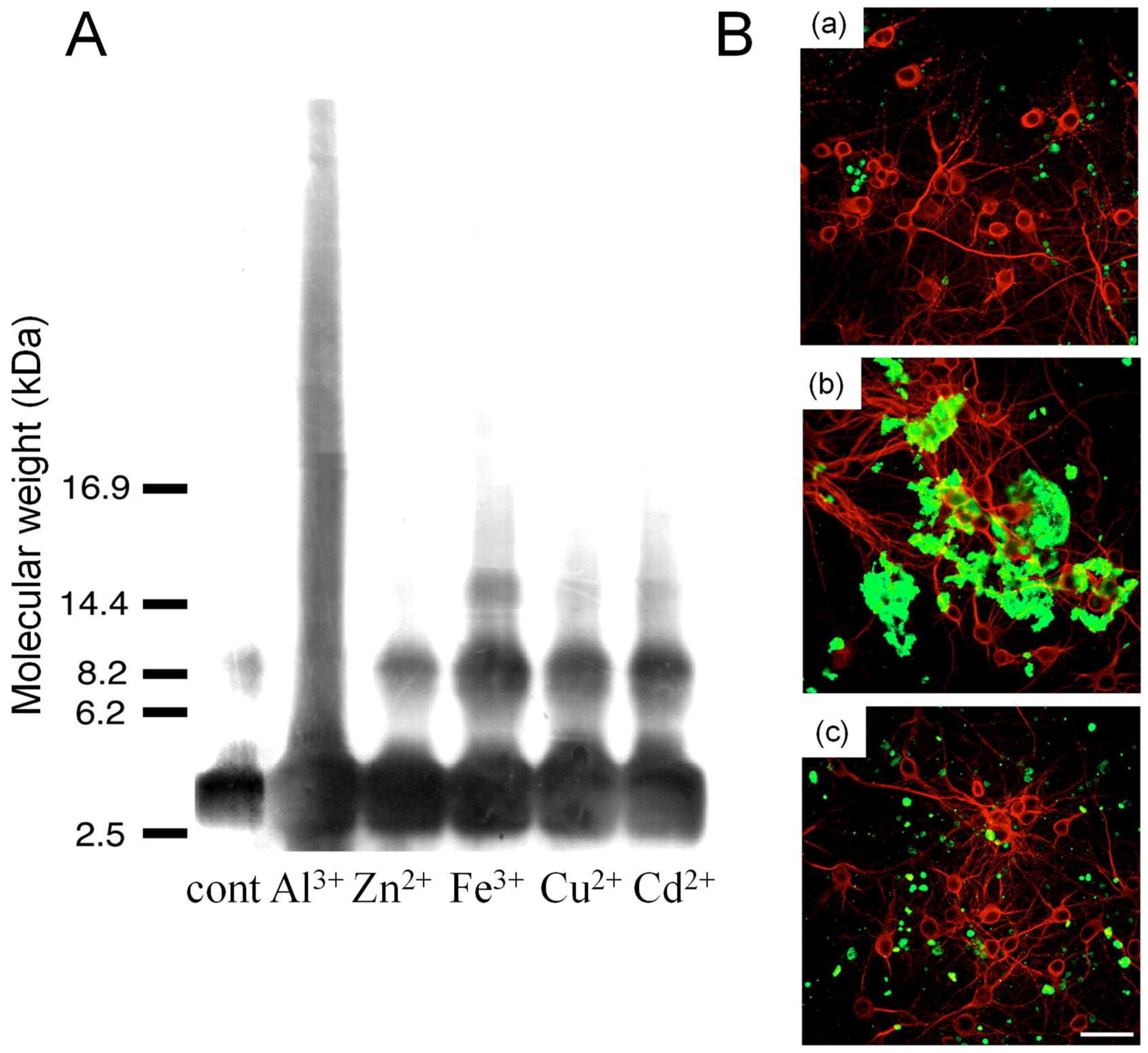
3.2. Metals and AβP
3.3. Isomerization and Racemization of Asp Residues in AβP
4. Prion Diseases
4.1. Pathogenesis of Prion Diseases and Prion Protein
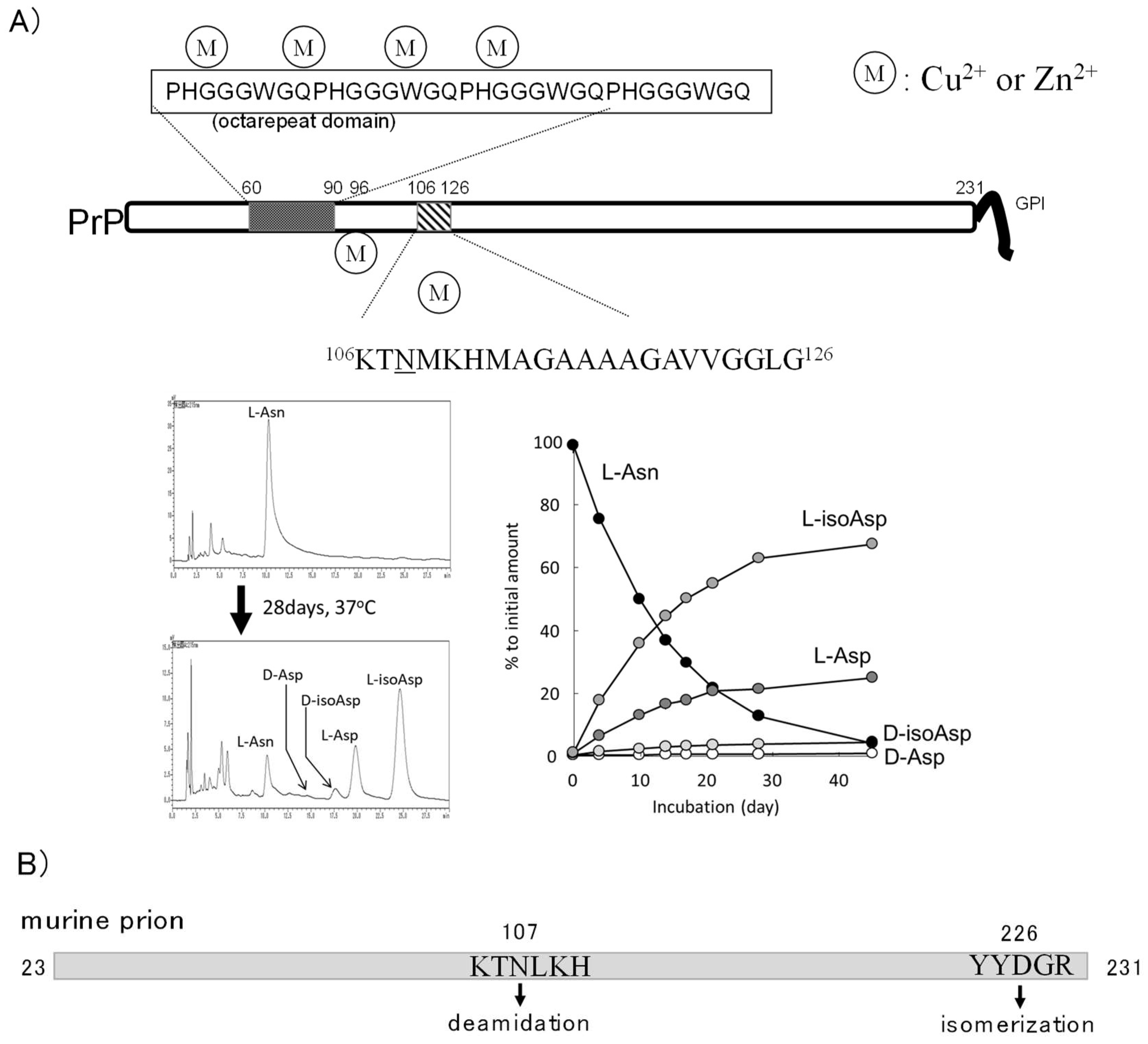
4.2. Prion Protein and Metals
4.3. Asn Deamidation in Prion Protein
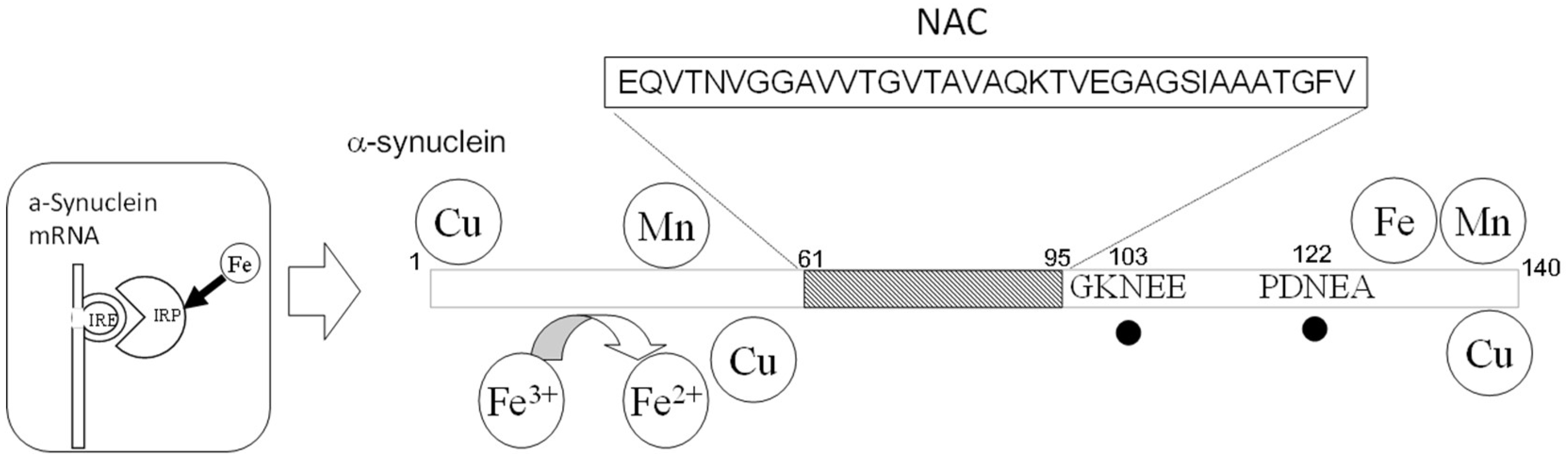
5. Lewy Body Diseases
6. Other Amyloidosis
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AβP | β-amyloid protein |
| AD | Alzheimer’s disease |
| AFM | Atomic force microscopy |
| ALS | Amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate |
| APP | Amyloid precursor protein |
| β2M | β2-Microglobulin |
| BSE | Bovine spongiform encephalopathy |
| CD | Far-UV circular dichroism |
| CJD | Creutzfeldt-Jakob disease |
| CSF | Cerebrospinal fluid |
| DLB | Dementia with Lewy bodies |
| FAP | Familial amyloid polyneuropathy |
| FT-ICR MS | Fourier transform ion cyclotron resonance mass spectrometry |
| GPI | Glycosylphosphatidylinositol |
| GSS | Sträussler-Scheinker syndrome |
| HPLC | High-performance liquid chromatography |
| IAPP | Islet amyloid polypeptide |
| IM-MS | Ion mobility mass spectrometry |
| IRE | Iron-responsive element |
| NAC | Non-amyloid component |
| NFT | Neurofibrillary tangles |
| NMDA | N-methyl-d-aspartate |
| PD | Parkinson’s disease |
| PIMT | Protein l-isoaspartyl O-methyltransferase |
| PrP | Prion protein |
| SOD | Superoxide dismutase |
| ZIP | Zrt-, Irt-like protein |
| ThT | Thioflavin T |
References
- Sipe, J.D.; Cohen, A.S. Review: History of the amyloid fibril. J. Struct. Biol. 2000, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Loo, D.; Mollee, P.N.; Renaut, P.; Hill, M.M. Proteomics in molecular diagnosis: Typing of amyloidosis. J. Biomed. Biotechnol. 2011, 754109. [Google Scholar] [CrossRef] [PubMed]
- Carrell, R.W.; Lomas, D.A. Conformational disease. Lancet 1997, 350, 134–138. [Google Scholar] [CrossRef]
- Kawahara, M.; Negishi-Kato, M.; Sadakane, Y. Calcium dyshomeostasis and neurotoxicity of Alzheimer’s β-amyloid protein. Expert. Rev. Neurother. 2009, 9, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Matusch, A.; Palm, C.; Salber, D.; Morton, K.A.; Becker, J.S. Bioimaging of metals in brain tissue by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) and metallomics. Metallomics 2010, 2, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato-Negishi, M.; Tanaka, K. Cross talk between neurometals and amyloidogenic proteins at the synapse and the pathogenesis of neurodegenerative diseases. Metallomics 2017, 9, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Muramoto, K.; Kobayashi, K.; Mori, H.; Kuroda, Y. Aluminum promotes the aggregation of Alzheimer’s amyroid β-protein in vitro. Biochem. Biophys. Res. Commun. 1994, 198, 531–535. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Kawahara, M. Aggregation of amyloid β-protein and its neurotoxicity: Enhancement by aluminum and other metals. Tohoku J. Exp. Med. 1994, 174, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Kato, M.; Kuroda, Y. Effects of aluminum on the neurotoxicity of primary cultured neurons and on the aggregation of β-amyloid protein. Brain Res. Bull. 2001, 55, 211–217. [Google Scholar] [CrossRef]
- Kawahara, M.; Koyama, H.; Nagata, T.; Sadakane, Y. Zinc, copper, and carnosine attenuate neurotoxicity of prion fragment PrP106–126. Metallomics 2011, 3, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Sesti, F.; Tsakiri, E.; Gorgoulis, V.G. Non-enzymatic post-translational protein modifications and proteostasis network deregulation in carcinogenesis. J. Prot. 2013, 92, 274–298. [Google Scholar] [CrossRef] [PubMed]
- Soskić, V.; Groebe, K.; Schrattenholz, A. Nonenzymatic posttranslational protein modifications in ageing. Exp. Gerontol. 2008, 43, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, Y.; Fujii, N.; Nakagomi, K. Determination of rate constants for β-linkage isomerization of three specific aspartyl residues in recombinant human αA-crystallin protein by reversed-phase HPLC. J. Chromatogr. B 2011, 879, 3240–3246. [Google Scholar] [CrossRef] [PubMed]
- Sadakane, Y.; Konoha, K.; Kawahara, M.; Nakagomi, K. Quantification of structural alterations of l-Asp and l-Asn residues in peptides related to neuronal diseases by reversed-phase high-performance liquid chromatography. Chem. Biodivers. 2010, 7, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E. Protein deamidation. Proc. Natl. Acad. Sci. USA. 2002, 99, 5283–5288. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Deamidation of human proteins. Proc. Natl. Acad. Sci. USA. 2001, 98, 12409–12413. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Clarke, S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J. Biol. Chem. 1987, 262, 785–794. [Google Scholar] [PubMed]
- Stephenson, R.C.; Clarke, S. Succinimide formation from aspartyl and asparaginyl peptides as a model for the spontaneous degradation of proteins. J. Biol. Chem. 1989, 264, 6164–6170. [Google Scholar] [PubMed]
- Tyler-Cross, R.; Schirch, V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J. Biol. Chem. 1991, 266, 22549–22556. [Google Scholar] [PubMed]
- Kossiakoff, A.A. Tertiary structure is a principal determinant to protein deamidation. Science 1988, 240, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Brennan, T.V.; Clarke, S. Effect of adjacent histidine and cysteine residues on the spontaneous degradation of asparaginyl- and aspartyl-containing peptides. Int. J. Pept. Protein. Res. 1995, 45, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S. Aging as war between chemical and biochemical processes: protein methylation and the recognition of age-damaged proteins for repair. Ageing Res. Rev. 2003, 2, 263–285. [Google Scholar] [CrossRef]
- Kim, E.; Lowenson, J.D.; MacLaren, D.C.; Clarke, S.; Young, S.G. Deficiency of a protein-repair enzyme results in the accumulation of altered proteins, retardation of growth, and fatal seizures in mice. Proc. Natl. Acad. Sci. USA. 1997, 94, 6132–6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reissner, K.J.; Paranandi, M.V.; Luc, T.M.; Doyle, H.A.; Mamula, M.J.; Lowenson, J.D.; Aswad, D.W. Synapsin I is a major endogenous substrate for protein l-isoaspartyl methyltransferase in mammalian brain. J. Biol. Chem. 2006, 281, 8389–8398. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.X.; Doyle, H.A.; Mamula, M.J.; Aswad, D.W. Protein repair in the brain, proteomic analysis of endogenous substrates for protein l-isoaspartyl methyltransferase in mouse brain. J. Biol. Chem. 2006, 281, 33802–33813. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.J.; Hall, D.; Carver, J.A. Deamidation of N76 in human γS-crystallin promotes dimer formation. Biochim. Biophys. Acta 2016, 1860, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Pande, A.; Mokhor, N.; Pande, J. Deamidation of human γS-crystallin increases attractive protein interactions: Implications for cataract. Biochemistry 2015, 54, 4890–4899. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Satoh, K.; Harada, K.; Ishibashi, Y. Simultaneous stereoinversion and isomerization at specific aspartic acid residues in alpha A-crystallin from human lens. J. Biochem. 1994, 116, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Fujii, N.; Sakaue, H.; Sasaki, H.; Fujii, N. A rapid, comprehensive liquid chromatography-mass spectrometry (LC-MS)-based survey of the Asp isomers in crystallins from human cataract lenses. J. Biol. Chem. 2012, 287, 39992–40002. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Fujii, N. Isomerization of Asp residues plays an important role in αA-crystallin dissociation. FEBS J. 2016, 283, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K. Isomerization of aspartyl residues in crystallins and its influence upon cataract. Biochim. Biophys. Acta 2016, 1860, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Langmack, E.L.; Aswad, D.W. Partial repair of deamidation-damaged calmodulin by protein carboxyl methyltransferase. J. Biol. Chem. 1987, 262, 12283–12287. [Google Scholar] [PubMed]
- Szymanska, G.L.; Leszyk, J.D.; O’Connor, C.M. Carboxyl methylation of deamidated calmodulin increases its stability in Xenopus oocyte cytoplasm. Implications for protein repair. J. Biol. Chem. 1998, 273, 28516–28523. [Google Scholar] [CrossRef] [PubMed]
- Curnis, F.; Longhi, R.; Crippa, L.; Cattaneo, A.; Dondossola, E.; Bachi, A.; Corti, A. Spontaneous formation of l-isoaspartate and gain of function in fibronectin. J. Biol. Chem. 2006, 281, 36466–36476. [Google Scholar] [CrossRef] [PubMed]
- Barbariga, M.; Curnis, F.; Spitaleri, A.; Andolfo, A.; Zucchelli, C.; Lazzaro, M.; Magnani, G.; Musco, G.; Corti, A.; Alessio, M. Oxidation-induced structural changes of ceruloplasmin foster NGR motif deamidation that promotes integrin binding and signaling. J. Biol. Chem. 2014, 289, 3736–3748. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Ohtsuka, I.; Yokoyama, S.; Kato-Negishi, M.; Sadakane, Y. Membrane incorporation, channel formation, and disruption of calcium homeostasis by Alzheimer’s β-amyloid protein. Int. J. Alzheimer’s Dis. 2011, 304583. [Google Scholar] [CrossRef]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, F.L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Baumkötter, F.; Schmidt, N.; Vargas, C.; Schilling, S.; Weber, R.; Wagner, K.; Fiedler, S.; Klug, W.; Radzimanowski, J.; Nickolaus, S.; et al. Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J. Neurosci. 2014, 34, 11159–11172. [Google Scholar] [CrossRef] [PubMed]
- Yankner, B.A.; Duffy, L.K.; Kirschner, D.A. Neurotropic and neurotoxic effects of amyloid β protein: Reversal by tachykinin neuropeptides. Science 1990, 250, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.J.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. In vitro aging of β-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991, 563, 311–314. [Google Scholar] [CrossRef]
- Simmons, L.K.; May, P.C.; Tomaselli, K.J.; Rydel, R.E.; Fuson, K.S.; Brigham, E.F.; Wright, S.; Lieberburg, I.; Becker, G.W.; Brems, D.N.; et al. Secondary structure of amyloid β peptide correlates with neurotoxic activity in vitro. Mol. Pharmacol. 1994, 45, 373–379. [Google Scholar] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Aβ oligomers- a decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Wirths, O.; Multhaup, G.; Bayer, T.A. A modified β-amyloid hypothesis: Intraneuronal accumulation of the β-amyloid peptide-the first step of a fatal cascade. J. Neurochem. 2004, 91, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, R.; Mizuno, T.; Mori, S.; Nakajima, K.; Fushiki, S.; Yanagisawa, K. Age-dependent change in the levels of Aβ40 and Aβ42 in cerebrospinal fluid from control subjects, and a decrease in the ratio of Aβ42 to Aβ40 level in cerebrospinal fluid from Alzheimer’s disease patients. Eur. Neurol. 2000, 43, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Asano, S.; Suwa, Y.; Morita, T.; Kataoka, K.; Mori, H.; Endo, N. Rifampicin prevents the aggregation and neurotoxicity of amyloid β protein in vitro. Biochem. Biophys. Res. Commun. 1994, 204, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Hasegawa, K.; Naiki, H.; Yamada, M. Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro. J. Neurosci. Res. 2004, 75, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Frazzini, V.; Silvestri, E.; Lattanzio, R.; La Sorda, R.; Piantelli, M.; Canzoniero, L.M.; Ciavardelli, D.; Rizzarelli, E.; Sensi, S.L. Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS ONE 2011, 6, e17971. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Nadackal, T.G.; Thomas, K. Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-β aggregation. Neuroreport 2001, 12, 3263–3267. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castaño, E.M.; Frangione, B. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Exley, C.; Price, N.C.; Kelly, S.M.; Birchall, J.D. An interaction of β-amyloid with aluminium in vitro. FEBS Lett. 1993, 324, 293–295. [Google Scholar] [CrossRef] [Green Version]
- Pratico, D.; Uryu, K.; Sung, S.; Tang, S.; Trojanowski, J.Q.; Lee, V.M. Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J. 2002, 16, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer Aβ amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Atwood, C.S.; Moir, R.D.; Huang, X.; Scarpa, R.C.; Bacarra, N.M.; Romano, D.M.; Hartshorn, M.A.; Tanzi, R.E.; Bush, A.I. Dramatic aggregation of Alzheimer aβ by Cu(II) is induced by conditions representing physiological acidosis. J. Biol. Chem. 1998, 273, 12817–12826. [Google Scholar] [CrossRef] [PubMed]
- Solomonov, I.; Korkotian, E.; Born, B.; Feldman, Y.; Bitler, A.; Rahimi, F.; Li, H.; Bitan, G.; Sagi, I. Zn2+-Aβ40 complexes form metastable quasi-spherical oligomers that are cytotoxic to cultured hippocampal neurons. J. Biol. Chem. 2012, 287, 20555–20564. [Google Scholar] [CrossRef] [PubMed]
- Dyrks, T.; Dyrks, E.; Hartmann, T.; Masters, C.; Beyreuther, K. Amyloidogenicity of βA4 and βA4-bearing amyloid protein precursor fragments by metal-catalyzed oxidation. J. Biol. Chem. 1992, 267, 18210–18217. [Google Scholar] [PubMed]
- Chen, W.T.; Liao, Y.H.; Yu, H.M.; Cheng, I.H.; Chen, Y.R. Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-beta stability, oligomerization, and aggregation: amyloid-beta destabilization promotes annular protofibril formation. J. Biol. Chem. 2011, 286, 9646–9656. [Google Scholar] [CrossRef] [PubMed]
- Bolognin, S.; Zatta, P.; Lorenzetto, E.; Valenti, M.T.; Buffelli, M. β-Amyloid-aluminum complex alters cytoskeletal stability and increases ROS production in cortical neurons. Neurochem. Int. 2013, 62, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Kim, J.; Mirica, L.M. The effect of Cu(2+) and Zn(2+) on the Aβ42 peptide aggregation and cellular toxicity. Metallomics 2013, 5, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Yumoto, S.; Kakimi, S.; Ohsaki, A.; Ishikawa, A. Demonstration of aluminum in amyloid fibers in the cores of senile plaques in the brains of patients with Alzheimer’s disease. J. Inorg. Biochem. 2009, 103, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Multhaup, G.; Maher, F.; Bellingham, S.; Camakaris, J.; Zheng, H.; Bush, A.I.; Beyreuther, K.; Masters, C.L.; Cappai, R. The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 1999, 19, 9170–9179. [Google Scholar] [CrossRef] [PubMed]
- Gerber, H.; Wu, F.; Dimitrov, M.; Garcia Osuna, G.M.; Fraering, P.C. Zinc and copper differentially modulate amyloid precursor protein processing by γ-secretase and amyloid-β peptide production. J. Biol. Chem. 2017, 292, 3751–3767. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.X.; Tsatsanis, A.; Lim, L.Q.; Adlard, P.A.; Bush, A.I.; Duce, J.A. β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS ONE 2014, 9, e114174. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5’-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Takio, K.; Ogawara, M.; Selkoe, D.J. Mass spectrometry of purified amyloid β protein in Alzheimer’s disease. J. Biol. Chem. 1992, 267, 17082–17086. [Google Scholar] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J.; et al. Structural alterations in the peptide backbone of β-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar] [PubMed]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Woods, A.S.; Cotter, R.J.; Gowing, E.; Ball, M.J. β-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc. Natl. Acad. Sci. USA. 1993, 90, 10836–10840. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Emmerling, M.R.; Woods, A.S.; Cotter, R.J.; Roher, A.E. Isolation, chemical characterization, and quantitation of Aβ 3-pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits. Biochem. Biophys. Res. Commun. 1997, 237, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Wakutani, Y.; Watanabe, K.; Adachi, Y.; Wada-Isoe, K.; Urakami, K.; Ninomiya, H.; Saido, T.C.; Hashimoto, T.; Iwatsubo, T.; Nakashima, K. Novel amyloid precursor protein gene missense mutation (D678N) in probable familial Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Hashimoto, T.; Wakutani, Y.; Urakami, K.; Nakashima, K.; Condron, M.M.; Tsubuki, S.; Saido, T.C.; Teplow, D.B.; Iwatsubo, T. The Tottori (D7N) and English (H6R) familial Alzheimer disease mutations accelerate Aβ fibril formation without increasing protofibril formation. J. Biol. Chem. 2007, 282, 4916–4923. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Condron, M.M.; Teplow, D.B. Effects of the English (H6R) and Tottori (D7N) familial Alzheimer disease mutations on amyloid β-protein assembly and toxicity. J. Biol. Chem. 2010, 285, 23186–23197. [Google Scholar] [CrossRef] [PubMed]
- Gessel, M.M.; Bernstein, S.; Kemper, M.; Teplow, D.B.; Bowers, M.T. Familial Alzheimer’s disease mutations differentially alter amyloid β-protein oligomerization. ACS Chem. Neurosci. 2012, 3, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.; Rebeck, G.W.; Greenberg, S.M. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann. Neurol. 2001, 49, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Tomidokoro, Y.; Rostagno, A.; Neubert, T.A.; Lu, Y.; Rebeck, G.W.; Frangione, B.; Greenberg, S.M.; Ghiso, J. Iowa variant of familial Alzheimer’s disease: accumulation of posttranslationally modified AβD23N in parenchymal and cerebrovascular amyloid deposits. Am. J. Pathol. 2010, 176, 1841–1854. [Google Scholar] [CrossRef] [PubMed]
- Fossati, S.; Todd, K.; Sotolongo, K.; Ghiso, J.; Rostagno, A. Differential contribution of isoaspartate post-translational modifications to the fibrillization and toxic properties of amyloid β and the Asn23 Iowa mutation. Biochem. J. 2013, 456, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, H.; Shimizu, T.; Nakajima, M.; Mori, H.; Shirasawa, T. Synthesis, aggregation, and neurotoxicity of the Alzheimer’s Aβ1-42 amyloid peptide and its isoaspartyl isomers. Bioorg. Med. Chem. Lett. 1999, 9, 953–956. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid β peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally applied synthetic peptide isoAsp7-Aβ(1–42) triggers cerebral β-amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Zatsepina, O.G.; Kechko, O.I.; Mitkevich, V.A.; Kozin, S.A.; Yurinskaya, M.M.; Vinokurov, M.G.; Serebryakova, M.V.; Rezvykh, A.P.; Evgen’ev, M.B.; Makarov, A.A. Amyloid-β with isomerized Asp7 cytotoxicity is coupled to protein phosphorylation. Sci. Rep. 2018, 8, 3518. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, T.; Asano, S.; Furiya, Y.; Shirasawa, T.; Endo, N.; Mori, H. Racemization of Asp23 residue affects the aggregation properties of Alzheimer amyloid β protein analogues. J. Biol. Chem. 1994, 269, 10205–10208. [Google Scholar] [PubMed]
- Sakai-Kato, K.; Naito, M.; Utsunomiya-Tate, N. Racemization of the amyloidal β Asp1 residue blocks the acceleration of fibril formation caused by racemization of the Asp23 residue. Biochem. Biophys. Res. Commun. 2007, 364, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Osaki, D.; Hiramatsu, H. Citrullination and deamidation affect aggregation properties of amyloid β-proteins. Amyloid 2016, 23, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prion diseases and the BSE crisis. Science 1997, 278, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Sandmeier, E.; Hunziker, P.; Kunz, B.; Sack, R.; Christen, P. Spontaneous deamidation and isomerization of Asn108 in prion peptide 106–126 and in full-length prion protein. Biochem. Biophys. Res. Commun. 1999, 261, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, D.; Koyama, H.; Ohkawara, S.; Sadakane, Y.; Kawahara, M. Involvement of trace elements in the pathogenesis of prion diseases. Curr. Pharam. Biotech. 2014, 15, 1049–1057. [Google Scholar] [CrossRef]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, N.; Herms, J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem. 2003, 86, 538–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, A.R.; Collins, S.J.; Maher, F.; Jobling, M.F.; Stewart, L.R.; Thyer, J.M.; Beyreuther, K.; Masters, C.L.; Cappai, R. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am. J. Pathol. 1999, 155, 1723–1730. [Google Scholar] [CrossRef]
- Gasperini, L.; Meneghetti, E.; Pastore, B.; Benetti, F.; Legname, G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid. Redox Signal. 2015, 22, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Alfaidy, N.; Chauvet, S.; Donadio-Andrei, S.; Salomon, A.; Saoudi, Y.; Richaud, P.; Aude-Garcia, C.; Hoffmann, P.; Andrieux, A.; Moulis, J.M.; et al. Prion protein expression and functional importance in developmental angiogenesis: Role in oxidative stress and copper homeostasis. Antioxid. Redox Signal. 2015, 18, 400–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, G.S.; Murray, I.; Hosszu, L.L.; Gibbs, N.; Waltho, J.P.; Clarke, A.R.; Collinge, J. Location and properties of metal-binding sites on the human prion protein. Proc. Natl. Acad. Sci. USA. 2001, 98, 8531–8535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt-Ulms, G.; Ehsani, S.; Watts, J.C.; Westaway, D.; Wille, H. Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS ONE 2009, 4, e7208. [Google Scholar] [CrossRef] [PubMed]
- Watt, N.T.; Griffiths, H.H.; Hooper, N.M. Neuronal zinc regulation and the prion protein. Prion 2013, 7, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimer’s Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Kong, Q.; Luo, X.; Petersen, R.B.; Meyerson, H.; Singh, N. Prion protein (PrP) knock-out mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS ONE 2009, 4, e6115. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, V.; Massignan, T.; Chiesa, R.; Morbin, M.; Mazzoleni, G.; Diomede, L.; Angeretti, N.; Colombo, L.; Forloni, G.; Tagliavini, F.; et al. Synthetic miniprion PrP106. J. Biol. Chem. 2002, 277, 31327–31334. [Google Scholar] [CrossRef] [PubMed]
- De Gioia, L.; Selvaggini, C.; Ghibaudi, E.; Diomede, L.; Bugiani, O.; Forloni, G.; Tagliavini, F.; Salmona, M. Conformational polymorphism of the amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. J. Biol. Chem. 1994, 269, 7859–7862. [Google Scholar] [PubMed]
- Grasso, D.; Milardi, D.; La Rosa, C.; Rizzarelli, E. The different role of Cu++ and Zn++ ions in affecting the interaction of prion peptide PrP106–126 with model membranes. Chem. Commun. 2004, 21, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Yang, D.S.; Yang, Y.; Chishti, M.A.; Meng, L.J.; Kretzschmar, H.A.; Yip, C.M.; Fraser, P.E.; Westaway, D. Copper(II)-induced conformational changes and protease resistance in recombinant and cellular PrP effect of protein age and deamidation. J. Biol. Chem. 2000, 275, 19121–19131. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kågedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benskey, M.J.; Perez, R.G.; Manfredsson, F.P. The contribution of alpha synuclein to neuronal survival and function-implications for Parkinson’s disease. J. Neurochem. 2016, 137, 331–359. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, M.L.; Schulze, S.E.; Lai, B.T.; Gray, H.B. Deamidation of alpha-synuclein. Protein Sci. 2009, 18, 1766–1773. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Angelova, D.M.; Jones, H.B.L.; Brown, D.R. Levels of α- and β-synuclein regulate cellular susceptibility to toxicity from α-synuclein oligomers. FASEB J. 2018, 32, 995–1006. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.M.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1790, 615–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, M.S. Human IAPP amyloidogenic properties and pancreatic β-cell death. Cell Calcium 2014, 56, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, M.F.; Sinopoli, A.; Pappalardo, G. On the environmental factors affecting the structural and cytotoxic properties of IAPP peptides. J. Diabetes Res. 2015, 918573. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.C.; Ha, E.; Singh, S.; Legesse, L.; Ahmad, S.; Karnaukhova, E.; Donaldson, R.P.; Jeremic, A.M. Copper(II)-human amylin complex protects pancreatic cells from amylin toxicity. Phys. Chem. Chem. Phys. 2013, 15, 12558–12571. [Google Scholar] [CrossRef] [PubMed]
- Hekman, C.M.; DeMond, W.S.; Kelley, P.J.; Mauch, S.F.; Williams, J.D. Isolation and identification of cyclic imide and deamidation products in heat stressed pramlintide injection drug product. J. Pharm. Biomed. Anal. 1999, 20, 763–772. [Google Scholar] [CrossRef]
- Dunkelberger, E.B.; Buchanan, L.E.; Marek, P.; Cao, P.; Raleigh, D.P.; Zanni, M.T. Deamidation accelerates amyloid formation and alters amylin fiber structure. J. Am. Chem. Soc. 2012, 134, 12658–12667. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.R.; Driscoll, M.; Raleigh, D.P. Low levels of asparagine deamidation can have a dramatic effect on aggregation of amyloidogenic peptides: implications for the study of amyloid formation. Protein Sci. 2002, 11, 342–349. [Google Scholar] [CrossRef] [PubMed]
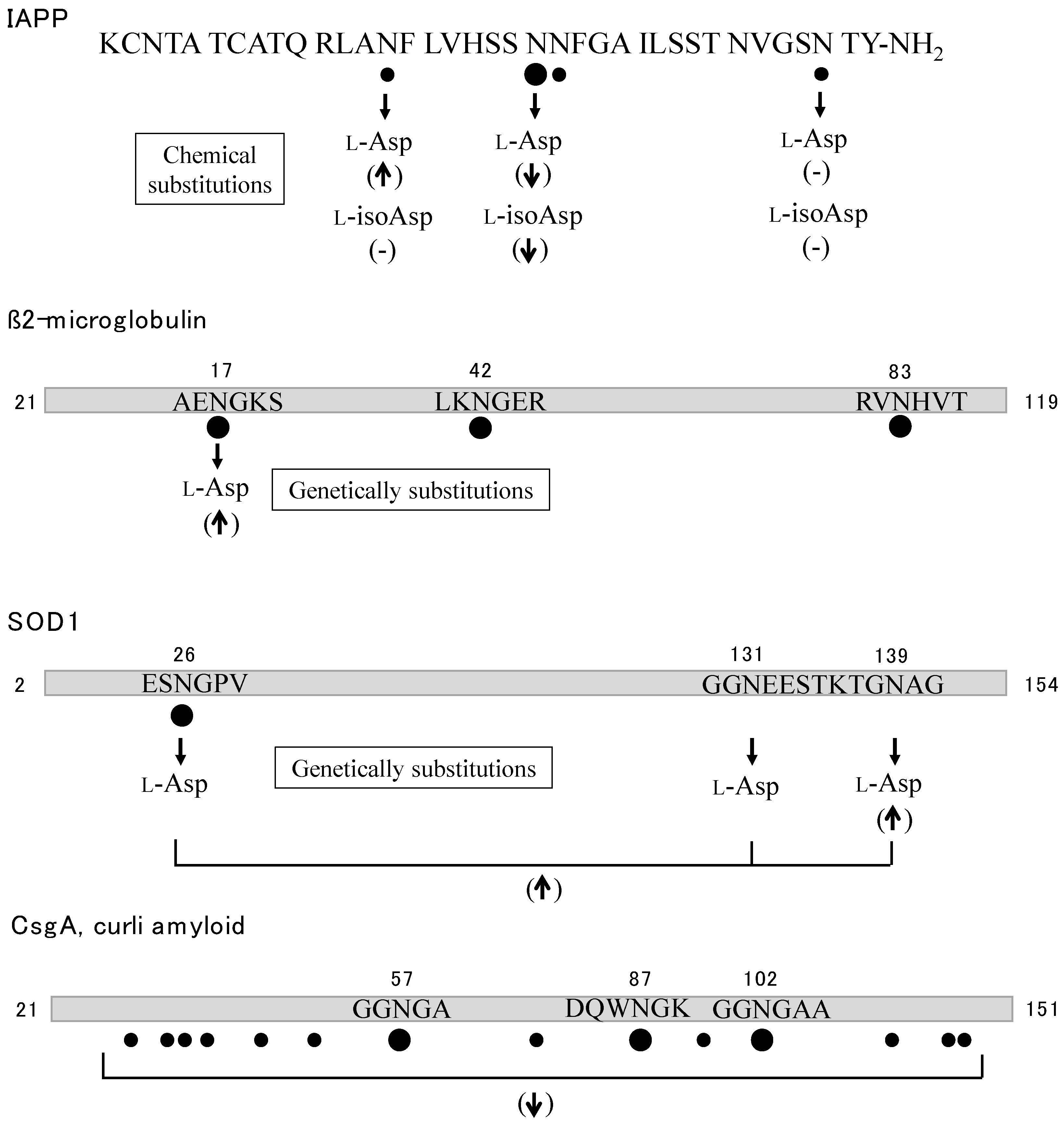
- Nguyen, P.T.; Zottig, X.; Sebastiao, M.; Bourgault, S. Role of Site-Specific Asparagine Deamidation in Islet Amyloid Polypeptide Amyloidogenesis: Key Contributions of Residues 14 and 21. Biochemistry 2017, 56, 3808–3817. [Google Scholar] [CrossRef] [PubMed]
- Odani, H.; Oyama, R.; Titani, K.; Ogawa, H.; Saito, A. Purification and complete amino acid sequence of novel β2-microglobulin. Biochem. Biophys. Res. Commun. 1990, 168, 1223–1229. [Google Scholar] [CrossRef]
- Li, X.; Yu, X.; Costello, C.E.; Lin, C.; O’Connor, P.B. Top-down study of β2-microglobulin deamidation. Anal. Chem. 2012, 84, 6150–6157. [Google Scholar] [CrossRef] [PubMed]
- Kad, N.M.; Thomson, N.H.; Smith, D.P.; Smith, D.A.; Radford, S.E. β2-Microglobulin and its deamidated variant, N17D form amyloid fibrils with a range of morphologies in vitro. J. Mol. Biol. 2001, 313, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Rhodes, N.R.; Abdolvahabi, A.; Kohn, T.; Cook, N.P.; Marti, A.A.; Shaw, B.F. Deamidation of asparagine to aspartate destabilizes Cu, Zn superoxide dismutase, accelerates fibrillization, and mirrors ALS-linked mutations. J. Am. Chem. Soc. 2013, 135, 15897–15908. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shu, Q.; Frieden, C.; Gross, M.L. Deamidation slows curli amyloid-protein aggregation. Biochemistry 2017, 56, 2865–2872. [Google Scholar] [CrossRef] [PubMed]
- Radford, S.E.; Gosal, W.S.; Platt, G.W. Towards an understanding of the structural molecular mechanism of β2-microglobulin amyloid formation in vitro. Biochim. Biophys. Acta 2005, 1753, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Joseph, C.A.; Borotto, N.B.; Gill, V.L.; Maroney, M.J.; Vachet, R.W. Unique effect of Cu(II) in the metal-induced amyloid formation of β-2-microglobulin. Biochemistry 2014, 53, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Netter, P.; Kessler, M.; Gaucher, A.; Burnel, D.; Fener, P. Aluminium and dialysis associated arthropathy. Nephron 1991, 59, 669. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, E.; Furukawa, Y. Copper homeostasis as a therapeutic target in amyotrophic lateral sclerosis with SOD1 mutations. Int. J. Mol. Sci. 2016, 17, 636. [Google Scholar] [CrossRef] [PubMed]
- Byström, R.; Andersen, P.M.; Gröbner, G.; Oliveberg, M. SOD1 mutations targeting surface hydrogen bonds promote amyotrophic lateral sclerosis without reducing apo-state stability. J. Biol. Chem. 2010, 285, 19544–19552. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.E.; Robinson, A.B. Prediction of protein deamidation rates from primary and three-dimensional structure. Proc. Natl. Acad. Sci. USA. 2001, 98, 4367–4372. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.L.; Chapman, M.R. Curli biogenesis: order out of disorder. Biochim. Biophys. Acta 2014, 1843, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, D.; Kawahara, M. Carnosine: A possible drug for vascular dementia. J. Vasc. Med. Surg. 2014, 2, 1–7. [Google Scholar]
- Kawahara, M.; Tanaka, K.I.; Kato-Negishi, M. Zinc, carnosine, and neurodegenerative diseases. Nutrients 2018, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, H.; Swinden, J.; Pierscionek, B.K.; Alany, R.G. Analytical and physicochemical characterisation of the senile cataract drug dipeptide β-alanyl-l-histidine (carnosine). J. Pharm. Biomed. Anal. 2015, 114, 241–246. [Google Scholar] [CrossRef] [PubMed]



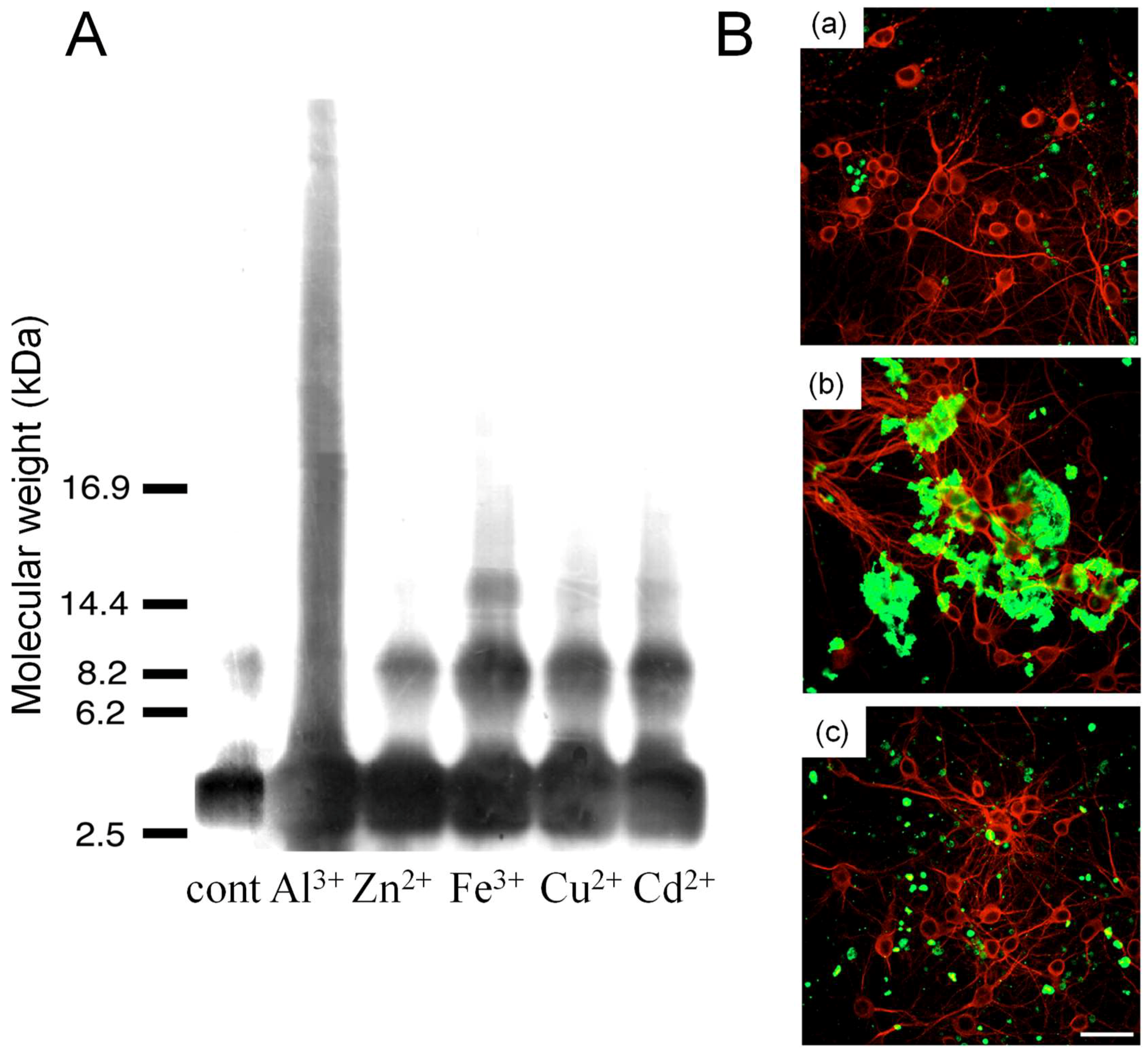




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Name Sequence | Binding Metals Structural Alteration of Asn or Asp | Functions of Amyloidogenic Proteins or Their Precursors |
|---|---|---|
| Alzheimer’s Disease AβP1–42 | Al, Zn, Cu, Fe Isomerization and racemization of Asp1 and Asp7 |
|
| DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA | ||
| Prion Diseases Prion protein; (PrP106–126) | Zn, Cu, Fe, Mn Deamidation of Asn108 |
|
| KKRPKPGGWNTGGSRYPGQGSPGGNRYPPQGGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQPHGGGWGQGGGTHSQWNKPSKPKTNMKHMAGAAAAGAVVGGLGGYMLGSAMSRPIIHFGSDYEDRYYRENMHRYPNQVYYRPMDEYSNQNNFVHDCVNITIKQHTVTTTTKGENFTETDVKMMERVVEQMCITQYERESQAYYQRGS | ||
| Lewy Body Diseases α-synuclein; (NAC, a fragment of α-synuclein) | Cu, Fe, Al Deamidation of Asn103 and Asn122 |
|
| MDVFMKGLSKAKEGVVAAAEKTKQGVAEAAGKTKEGVLYVGSKTKEGVVHGVTTVAEKTKEQVSNVGGAVVTGVTAVAHKTVEGAGNFAAATGLVKKDQKNESGFGPEGTMENSENMPVNPNNETYEMPPEEEYQDYDPEA | ||
| Type 2 Diabetes Islet amyloid peptide (IAPP, amylin) | Cu, Zn Deamidation of Asn21 |
|
| KCNTATCATQRLANFLVHSSNNFGAILSSTNVGSNTY | ||
| Dialysis Amyloidosis β2-microglobulin | Al, Cu, Zn, Ni Deamidation of Asn17, Asn42 and Asn83 |
|
| IQRTPKIQVYSRHPAENGKSNFLNCYVSGFHPSDIEVDLLKNGERIEKVEHSDLSFSKDWSFYLLYYTEFTPTEKDEYACRVNHVTLSQPKIVKWDRDM | ||
| Amyotropic Lateral Disorder (ALS) Cu, Zn-SOD1 | Cu, Zn Deamidation of Asn26 |
|
| ATKAVCVLKGDGPVQGIINFEQKESNGPVKVWGSIKGLTEGLHGFHVHEFGDNTAGCTSAGPHFNPLSRKHGGPKDEERHVGDLGNVTADKDGVADVSIEDSVISLSGDHCIIGRTLVVHEKADDLGKGGNEESTKTGNAGSRLACGVIGIAQ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadakane, Y.; Kawahara, M. Implications of Metal Binding and Asparagine Deamidation for Amyloid Formation. Int. J. Mol. Sci. 2018, 19, 2449. https://doi.org/10.3390/ijms19082449
Sadakane Y, Kawahara M. Implications of Metal Binding and Asparagine Deamidation for Amyloid Formation. International Journal of Molecular Sciences. 2018; 19(8):2449. https://doi.org/10.3390/ijms19082449
Chicago/Turabian StyleSadakane, Yutaka, and Masahiro Kawahara. 2018. "Implications of Metal Binding and Asparagine Deamidation for Amyloid Formation" International Journal of Molecular Sciences 19, no. 8: 2449. https://doi.org/10.3390/ijms19082449
APA StyleSadakane, Y., & Kawahara, M. (2018). Implications of Metal Binding and Asparagine Deamidation for Amyloid Formation. International Journal of Molecular Sciences, 19(8), 2449. https://doi.org/10.3390/ijms19082449