Genotyping-by-Sequencing Enhances Genetic Diversity Analysis of Crested Wheatgrass [Agropyron cristatum (L.) Gaertn.]




Abstract
:1. Introduction
2. Results
2.1. SNP Discovery and Characterization
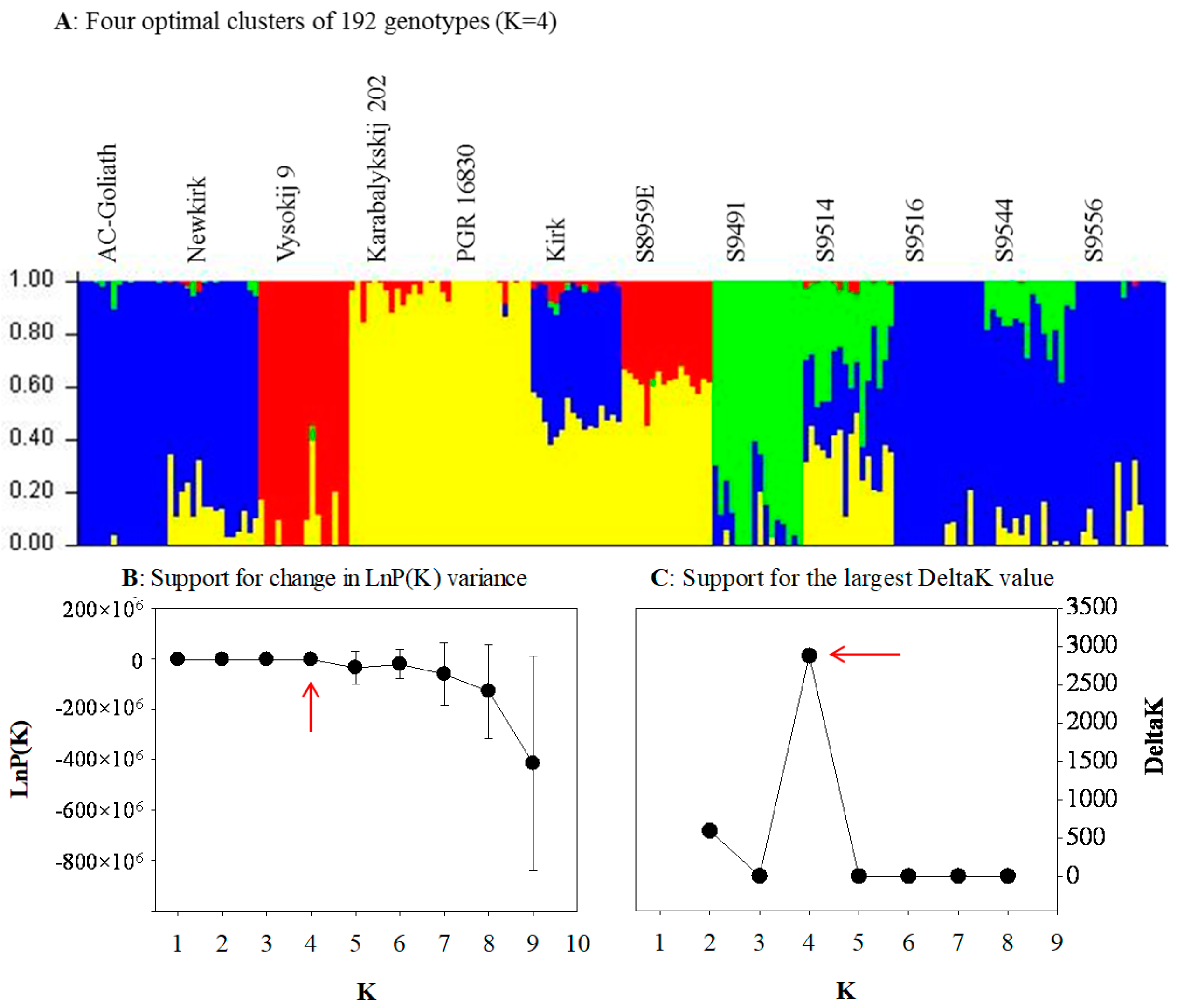
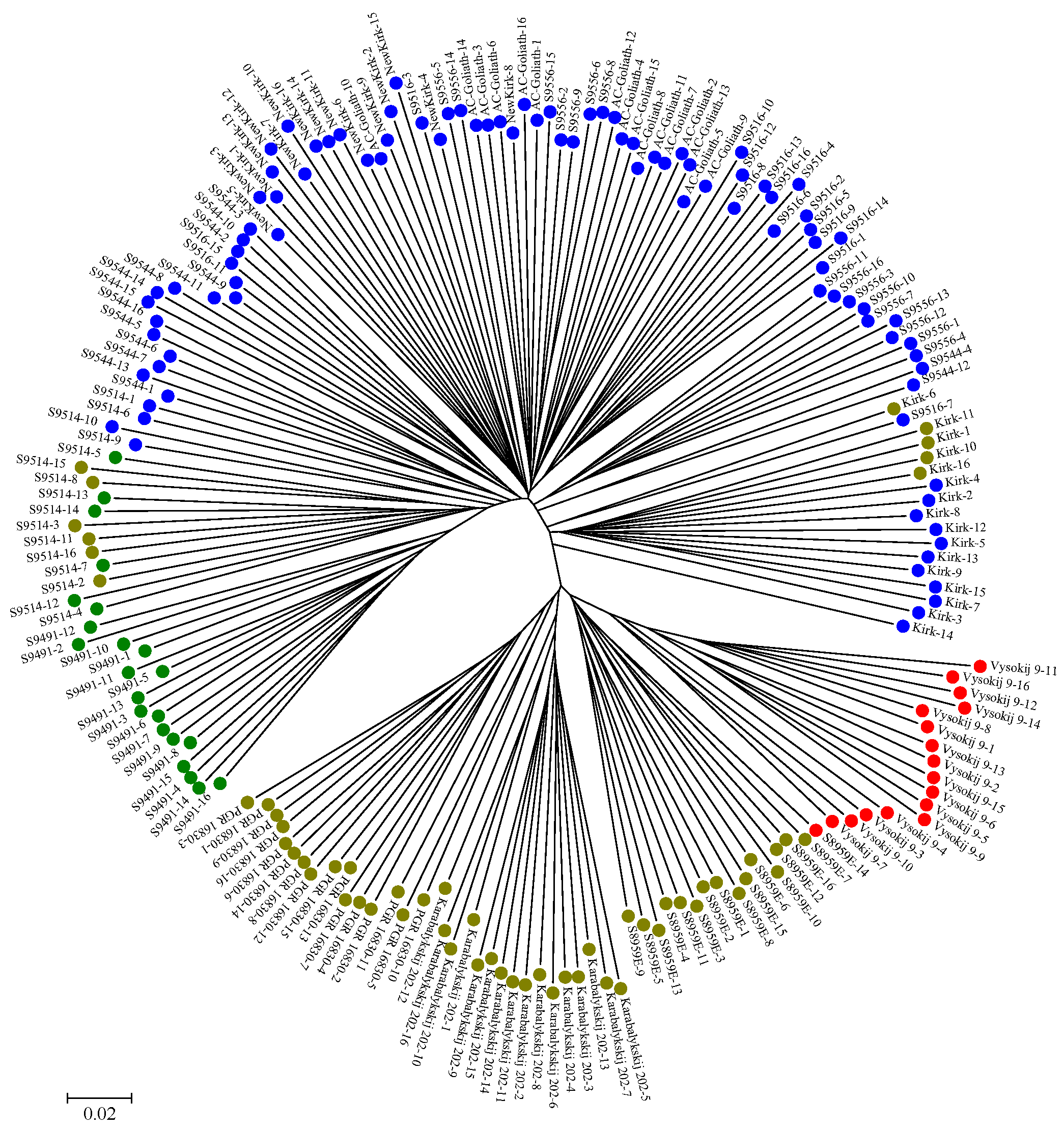
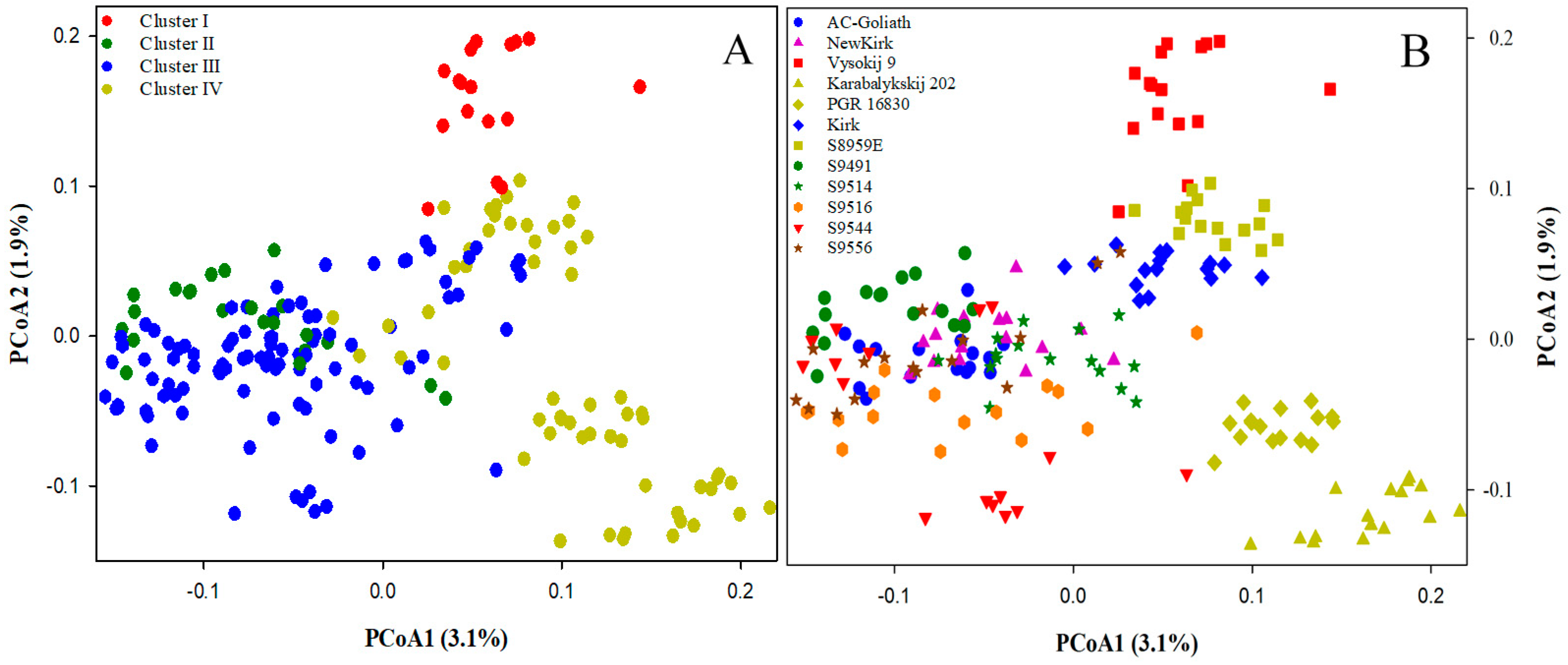
2.2. Genetic Structure and Relationship
2.3. Genetic Differentiation
2.4. Effects of Missing Data on Diversity Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Genotyping-by-Sequencing
4.3. Bioinformatics Analysis
4.4. Genetic Diversity Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fu, Y.B.; Peterson, G.W. Genetic diversity analysis with 454 pyrosequencing and genomic reduction confirmed the eastern and western division in the cultivated barley gene pool. Plant Genome 2011, 4, 226–237. [Google Scholar] [CrossRef]
- Peterson, B.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, G.W.; Dong, Y.; Horbach, C.; Fu, Y.B. Genotyping-by-sequencing for plant genetic diversity analysis: A lab guide for SNP genotyping. Diversity 2014, 6, 665–680. [Google Scholar] [CrossRef]
- Poland, J.A.; Rife, T.W. Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Poland, J.A.; Wight, C.P.; Jackson, E.W.; Tinker, N.A. Using genotyping-by-sequencing (GBS) for genomic discovery in cultivated oat. PLoS ONE 2014, 9, e102448. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Peterson, G.W.; Dong, Y. Increasing genome sampling and improving SNP genotyping for genotyping-by-sequencing with new combinations of restriction enzymes. G3 2016, 6, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.B.; Yang, M.H. Genotyping-by-sequencing and its application to oat genomic research. In Oat—Methods and Prtocols; Gasparis, S., Ed.; Springer Science+Business Media: New York, NY, USA, 2017; pp. 169–187. [Google Scholar] [CrossRef]
- Tinker, N.A.; Bekele, W.A.; Hattori, J. Haplotag: Software for haplotype-based genotyping-by-sequencing analysis. G3 2016, 6, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bekele, W.A.; Wight, C.P.; Peng, Y.; Langdon, T.; Latta, R.G.; Fu, Y.B.; Diederichsen, A.; Howarth, C.J.; Jellen, E.N.; et al. High-density markers profiling confirms ancestral genomes of Avena species and identifies D-genome chromosomes of hexaploid oat. Theor. Appl. Genet. 2016, 129, 2133–2149. [Google Scholar] [CrossRef] [PubMed]
- Bekele, W.A.; Wight, C.P.; Chao, S.; Howarth, C.J.; Tinker, N.A. Haplotype based genotyping-by-sequencing in oat genome research. Plant Biotechnol. J. 2018, 16, 1452–1463. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajaj, N.; Peterson, G.W.; Horbach, C.; Al-Shamaa, K.; Tinker, N.A.; Fu, Y.B. Genotyping-by-sequencing empowered genetic diversity analysis of Jordanian oat wild relative Avena sterilis. Genet. Resour. Crop Evol. [CrossRef]
- Li, P.; Bhattarai, S.; Peterson, G.P.; Coulman, B.E.; Schellenberg, M.P.; Biligetu, B.; Fu, Y.B. Genetic diversity of northern wheatgrass (Elymus lanceolatus ssp. lanceolatus) as revealed by genotyping-by-sequencing. Diversity 2018, 10, 23. [Google Scholar] [CrossRef]
- Dewey, D.R. The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. In Gene Manipulation in Plant Improvement, Proceedings of the 6th Stadler Genetics Symposium; Gustafson, J.P., Ed.; Columbia University Press: New York, NY, USA, 1984; pp. 209–279. [Google Scholar]
- Asay, K.H.; Jensen, K.B.; Hsiao, C.; Dewey, D.R. Probable origin of standard crested wheatgrass, Agropyron desertorum Fisch Ex Link, Schultes. Can. J. Plant Sci. 1992, 72, 763–772. [Google Scholar] [CrossRef]
- Rogler, G.A.; Lorenz, R.L. Crested wheatgrass-early history in the United States. J. Range Manag. 1983, 36, 91–93. [Google Scholar] [CrossRef]
- Chen, S.Y.; Ma, X.; Zhang, X.Q.; Huang, L.K.; Zhou, J.N. Genetic diversity and relationships among lines of five crested wheatgrass species (Poaceae: Agropyron) based on gliadin analysis. Genet. Mol. Res. 2013, 12, 5704–5713. [Google Scholar] [CrossRef] [PubMed]
- Mellish, A.; Coulman, B.E.; Ferdinandez, Y. Genetic relationships among selected crested wheatgrass cultivars and species determined on the basis of AFLP markers. Crop Sci. 2002, 42, 1662–1668. [Google Scholar] [CrossRef]
- Looman, J.; Heinrichs, D. Stability of crested wheatgrass pastures under long-term pasture use. Can. J. Plant Sci. 1973, 53, 501–506. [Google Scholar] [CrossRef]
- Asay, K.H.; Jensen, K.B. Wheatgrass. In Cool-Season Forage Grasses; Moser, L.E., Buxton, D., Casler, M.D., Eds.; Agron Monogr ASA, CSSA, SSSA: Madison, WI, USA, 1996; pp. 691–724. [Google Scholar]
- Zlatnik, E. Agropyron cristatum. In Fire Effects Information System, [Online]. U.S. Department of Agriculture, Forest Service, Rocky Mountain Research Station, Fire Sciences Laboratory (Producer), 1999; p. 8. Available online: https://www.fs.fed.us /database/feis/plants/graminoid/agrcri/all.html (accessed on 24 July 2018).
- Hull, G.J.; Klomp, A.C. Longevity of crested wheatgrass in the sagebrush-grass type in southern Idaho. J. Range Manag. 1966, 19, 5–11. [Google Scholar] [CrossRef]
- Sharma, H.C.; Gill, B.S.; Uyemoto, J.K. High levels of resistance in Agropyron species to barley yellow dwarf and wheat streak mosaic viruses. J. Phytopath. 1984, 110, 143–147. [Google Scholar] [CrossRef]
- Dong, Y.S.; Zhou, R.H.; Xu, S.J.; Li, L.H.; Cauderon, Y.; Wang, R.R. Desirable characteristics in perennial Triticeae collected in China for wheat improvement. Hereditas 1992, 116, 175–178. [Google Scholar] [CrossRef]
- Wu, J.; Yang, X.M.; Wang, H.; Li, H.J.; Li, L.H.; Li, X.Q.; Liu, W.H. The introgression of chromosome 6P specifying for increased numbers of florets and kernels from Agropyron cristatum into wheat. Theor. Appl. Genet. 2006, 114, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, V.; Madrid, E.; Said, M.; Rubiales, D.; Cabrera, A. Molecular and cytogenetic characterization of a common wheat-Agropyron cristatum chromosome translocation conferring resistance to leaf rust. Euphytica 2015, 201, 89–95. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Han, H.; Song, L.; Bai, L.; Gao, Z.; Zhang, Y.; Yang, X.; Gao, L.A.; Li, L. De novo transcriptome sequencing of Agropyron cristatum to identify available gene resources for the enhancement of wheat. Genomics 2015, 106, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Biligetu, B.; Coulman, B.E.; Schellenberg, M.P.; Fu, Y.B. RNA-Seq analysis of gene expression for floral development in crested wheatgrass (Agropyron cristatum L.). PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Biligetu, B.; Coulman, B.E.; Schellenberg, M.P.; Fu, Y.B. RNA-Seq analysis of plant maturity in crested wheatgrass (Agropyron cristatum L.). Genes. 2017, 8, 291. [Google Scholar] [CrossRef] [PubMed]
- Forster, J.W.; Jones, E.S.; Kölliker, R.; Drayton, M.C.; Dumsday, J.; Dupal, M.P.; Guthridge, K.M.; Mahoney, N.L.; van Zijil de Jong, E.; Smith, K.F. Development and implementation of molecular markers for forage crop improvement. In Molecular Breeding of Forage Crops; Spangenberg, G., Ed.; Kluwer Academic Press: Dordrecht, The Netherlands, 2001; pp. 101–133. [Google Scholar]
- Che, Y.H.; Li, H.J.; Yang, Y.P.; Yang, X.M.; Li, X.Q.; Li, L.H. On the use of SSR markers for the genetic characterization of the Agropyron cristatum (L.) Gaertn. in Northern China. Genet. Resour. Crop Evol. 2008, 55, 389–396. [Google Scholar] [CrossRef]
- Rogers, D.L.; Montalvo, A.M. Genetically Appropriate Choices for Plant materials to Maintain Biological Diversity; Report to the USDA Forest Service; University of California: Rocky Mountain Region, Lakewood, CO, USA, 2004; p. 343. [Google Scholar]
- Che, Y.H.; Yang, Y.P.; Yang, X.M.; Li, X.Q.; Li, L.H. Genetic diversity between ex situ and in situ samples of Agropyron cristatum (L.) Gaertn. based on simple sequence repeat molecular markers. Crop Past. Sci. 2011, 62, 639–644. [Google Scholar] [CrossRef]
- Che, Y.H.; Yang, Y.P.; Yang, X.M.; Li, X.Q.; Li, L.H. Phylogenetic relationship and diversity among Agropyron Gaertn. germplasm using SSRs markers. Plant Syst. Evol. 2015, 301, 163–170. [Google Scholar] [CrossRef]
- Lu, F.; Lipka, A.E.; Glaubitz, J.; Elshire, R.; Cherney, J.H.; Casler, M.D.; Buckler, E.S.; Costich, D.E. Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network-based SNP discovery protocol. PLoS Genet. 2013, 9, e1003215. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Stephens, M.; Donnelly, P. Influence of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Hamrick, J.L.; Godt, M.J.W. Allozyme diversity in plant species. In Plant Population Genetics, Breeding and Genetic Resources; Brown, A.H.D., Clegg, M.T., Kahler, A.L., Weir, B.S., Eds.; Sinauer Associates: Sunderland, MA, USA, 1989; pp. 43–63. [Google Scholar]
- Fu, Y.B.; Coulman, B.E.; Ferdinandez, Y.S.N.; Cayouette, J.; Peterson, P.M. Genetic diversity of fringed brome (Bromus ciliatus) as determined by amplified fragment length polymorphism. Can. J. Bot. 2005, 83, 1322–1328. [Google Scholar] [CrossRef]
- Biligetu, B.; Schellenberg, M.P.; Fu, Y.B. Detecting genetic diversity of side-oats grama grass populations using AFLP Marker. Can. J. Plant Sci. 2013, 93, 1105–1114. [Google Scholar] [CrossRef]
- Fu, Y.B.; Phan, A.T.; Coulman, B.E.; Richards, K.W. Genetic diversity in natural populations and corresponding seed collections of little bluestem as revealed by AFLP markers. Crop Sci. 2004, 44, 2254–2260. [Google Scholar] [CrossRef]
- Li, P.; Biligetu, B.; Coulman, B.E.; Schellenberg, M.P.; Fu, Y.B. Genotyping-by-sequencing data of 272 crested wheatgrass (Agropyron cristatum) genotypes. Data Brief 2017, 15, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Dominant markers and null alleles. Mol. Ecol. Notes 2007, 7, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Fu, Y.B. AveDissR: An R function for assessing genetic distinctness and genetic redundancy. Appl. Plant Sci. 2017, 5, 1700018. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; ISBN 3-900051-07-0. Available online: http://www.r-project.org/ (accessed on 3 August 2018).
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5. 5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Weir, B.S.; Hill, W.G. Estimating F-statistics. Ann. Rev. Genet. 2002, 36, 721–775. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lines | CN Number a | Alternative Identification a | Origin | Type |
|---|---|---|---|---|
| Kirk | CN108662 | PI 536010 | Canada | Cultivar |
| AC-Goliath | CN108673 | Canada | Cultivar | |
| NewKirk | FOR552 | Canada | Cultivar | |
| Vysokij 9 | CN30995 | PI 370654 | Siberia, Former Soviet Union, Omsk region | Genebank line |
| Karabalykskij 202 | CN31068 | PI 326204 | Kazakhstan, Former Soviet Union, Kustanai region | Genebank line |
| PGR 16830 | CN43478 | Kazakhstan | Genebank line | |
| S8959E | FOR917 | Siberia/Canada | Breeding line | |
| S9491 | S9491 | Canada | Breeding line | |
| S9514 | S9514 | Canada | Breeding line | |
| S9516 | S9516 | Canada | Breeding line | |
| S9544 | S9544 | Canada | Breeding line | |
| S9556 | S9556 | Canada | Breeding line |
| Model/Source of Variation | df | Sum of Squares | Variance Explained | Variance (%) a |
|---|---|---|---|---|
| 12 lines | ||||
| Among lines | 11 | 101,048.8 | 246.0 | 15.8 |
| Within lines | 372 | 488,598.0 | 1313.4 | 84.2 |
| Four clusters from STRUCTURE | ||||
| Among clusters | 3 | 54,736.5 | 193.3 | 12.1 |
| Within clusters | 380 | 534,910.3 | 1407.7 | 87.9 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baral, K.; Coulman, B.; Biligetu, B.; Fu, Y.-B. Genotyping-by-Sequencing Enhances Genetic Diversity Analysis of Crested Wheatgrass [Agropyron cristatum (L.) Gaertn.]. Int. J. Mol. Sci. 2018, 19, 2587. https://doi.org/10.3390/ijms19092587
Baral K, Coulman B, Biligetu B, Fu Y-B. Genotyping-by-Sequencing Enhances Genetic Diversity Analysis of Crested Wheatgrass [Agropyron cristatum (L.) Gaertn.]. International Journal of Molecular Sciences. 2018; 19(9):2587. https://doi.org/10.3390/ijms19092587
Chicago/Turabian StyleBaral, Kiran, Bruce Coulman, Bill Biligetu, and Yong-Bi Fu. 2018. "Genotyping-by-Sequencing Enhances Genetic Diversity Analysis of Crested Wheatgrass [Agropyron cristatum (L.) Gaertn.]" International Journal of Molecular Sciences 19, no. 9: 2587. https://doi.org/10.3390/ijms19092587
APA StyleBaral, K., Coulman, B., Biligetu, B., & Fu, Y.-B. (2018). Genotyping-by-Sequencing Enhances Genetic Diversity Analysis of Crested Wheatgrass [Agropyron cristatum (L.) Gaertn.]. International Journal of Molecular Sciences, 19(9), 2587. https://doi.org/10.3390/ijms19092587