Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling



{kind=link}
{kind=link}
{kind=link}
Abstract
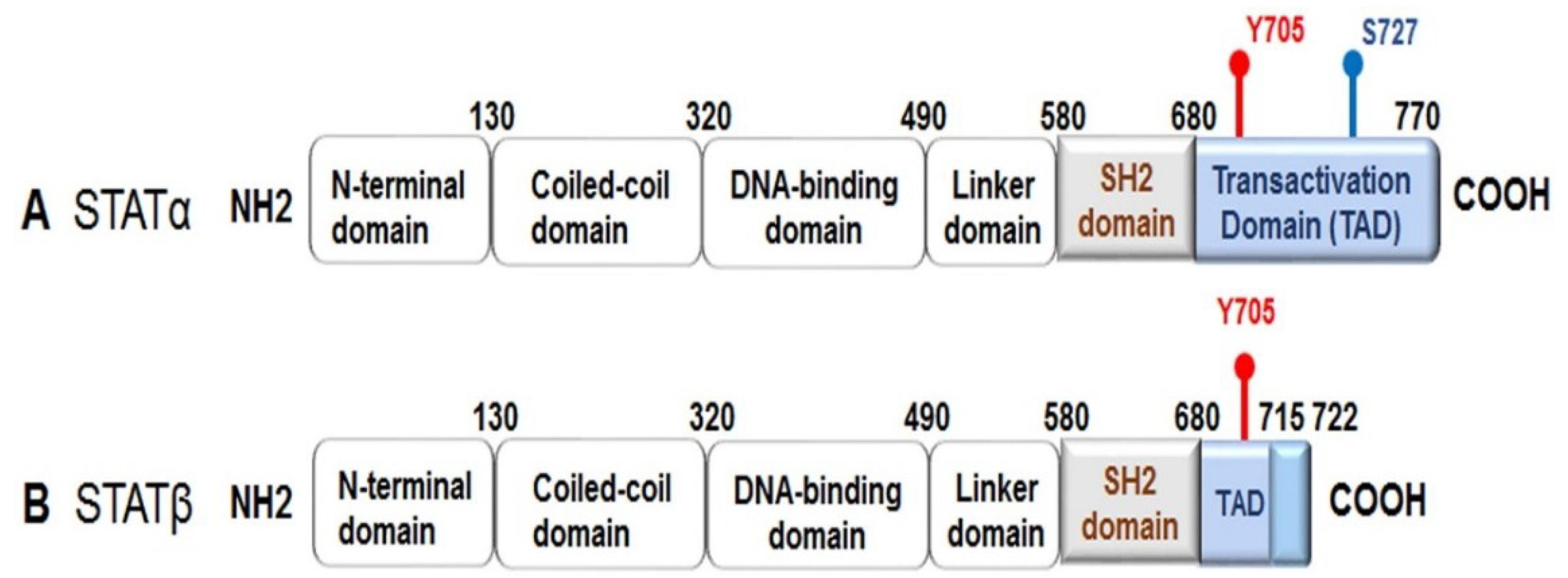
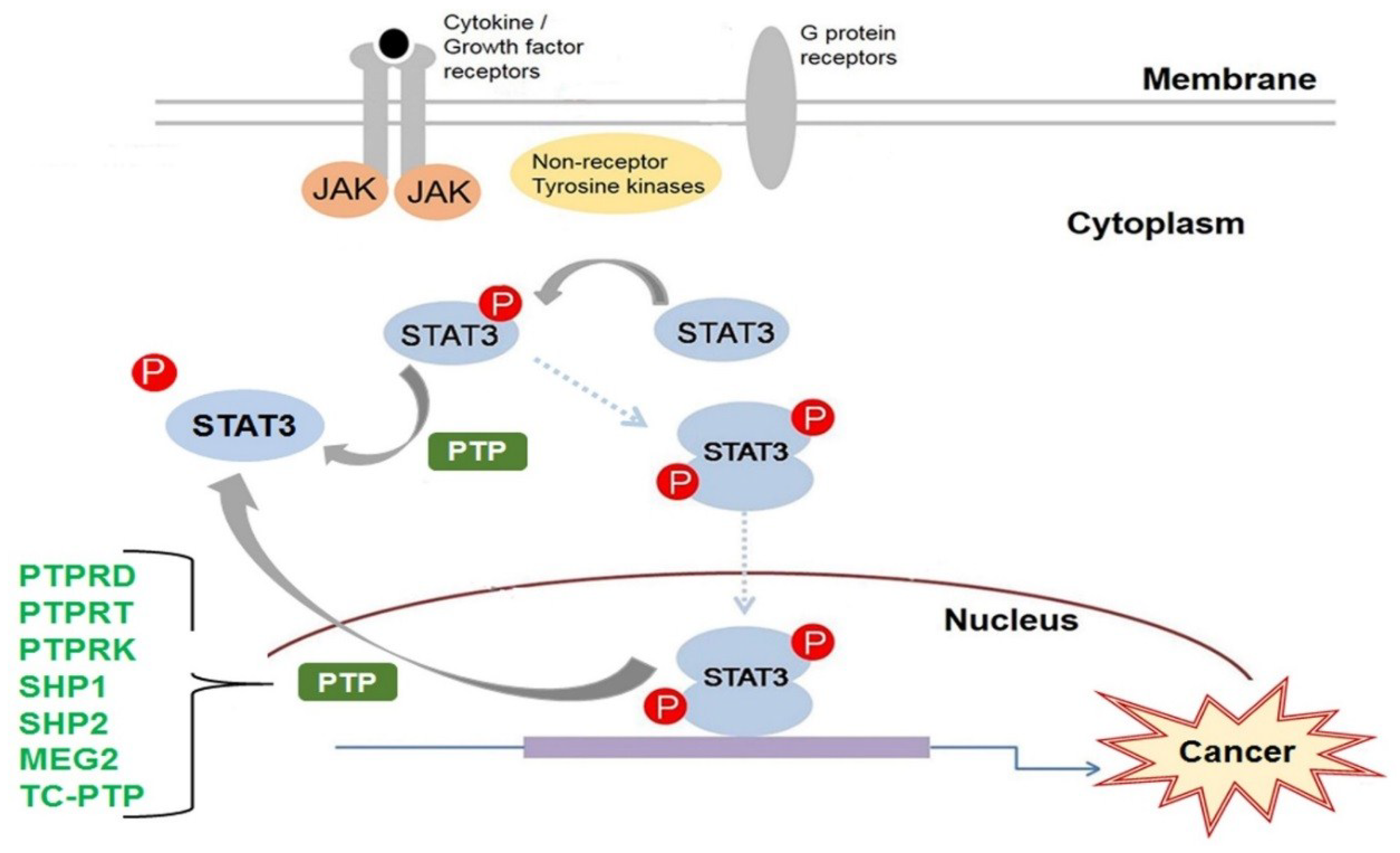
:1. Introduction
2. Protein Tyrosine Phosphatase Receptor-Type D
3. Protein Tyrosine Phosphatase Receptor-Type T
4. Protein Tyrosine Phosphatase Receptor-Type K
5. Src Homology Region 2 Domain-Containing Phosphatase 1
6. Src Homology Region 2 Domain-Containing Phosphatase-2
7. MEG2/Protein Tyrosine Phosphatase Non-Receptor Type 9
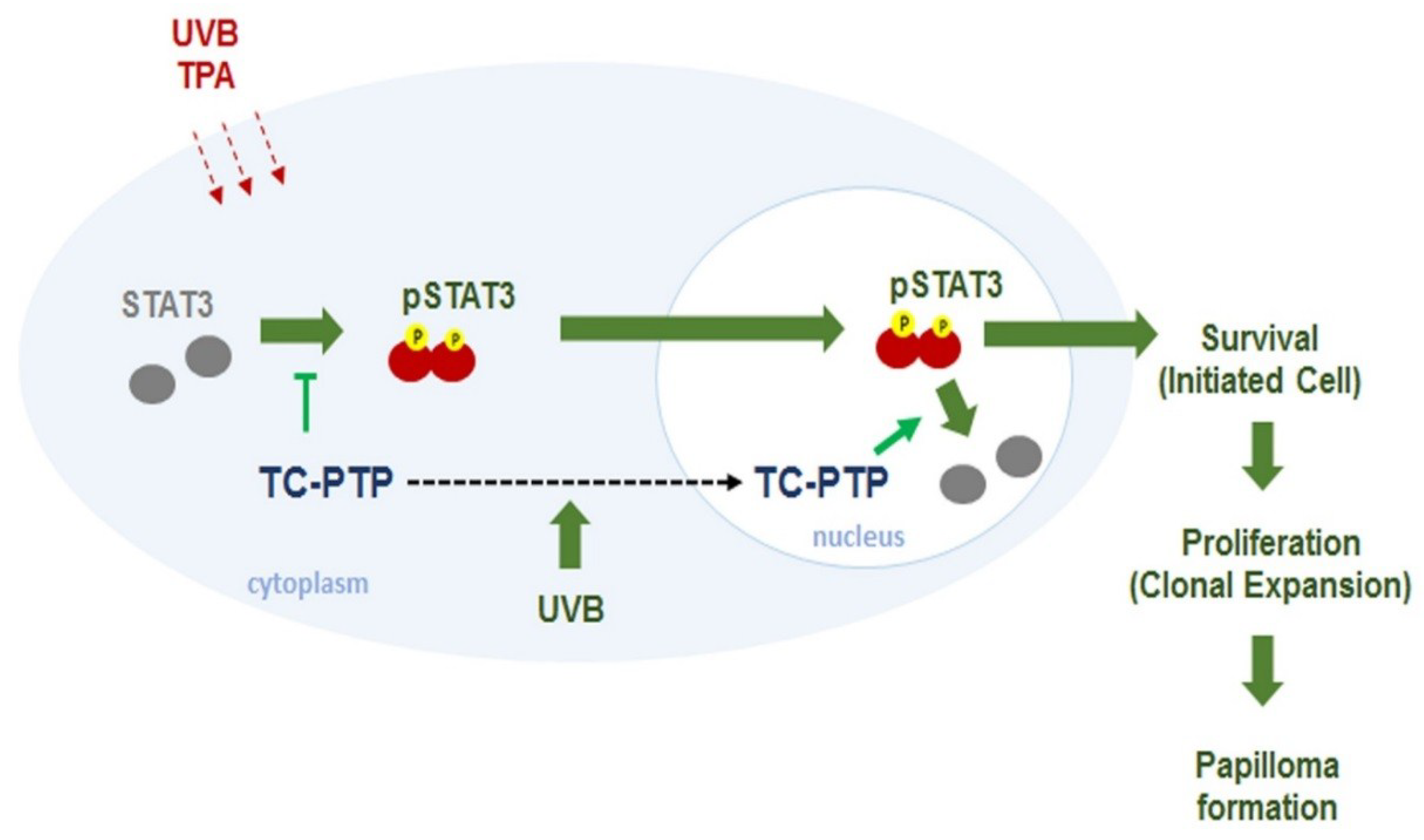
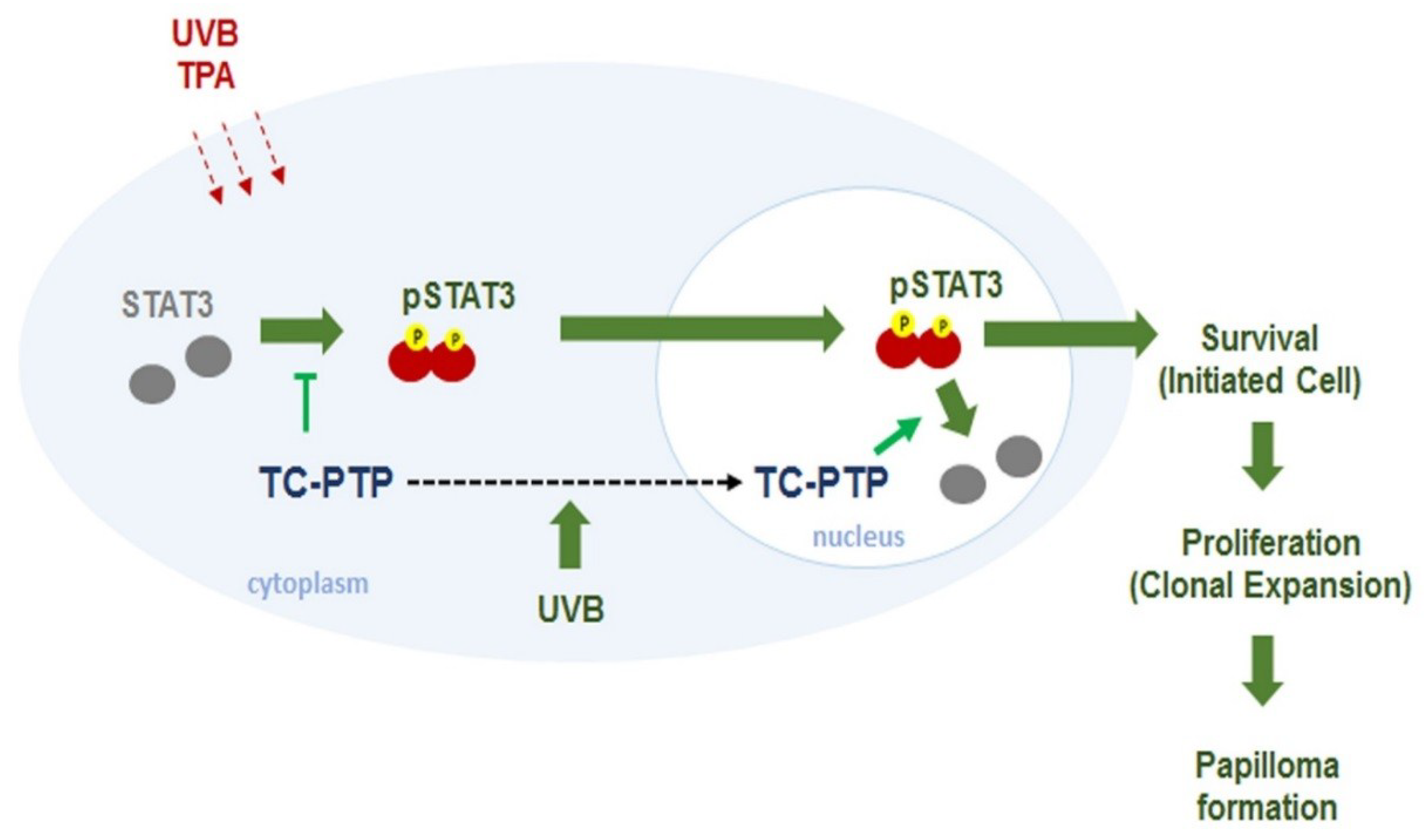
8. T-Cell Protein Tyrosine Phosphatase
9. Conclusions
Funding
Conflicts of Interest
References
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromberg, J. STAT proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Lee, C.K. What does STAT3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Caldenhoven, E.; van Dijk, T.B.; Solari, R.; Armstrong, J.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L.; de Groot, R.P. STAT3β, a splice variant of transcription factor STAT3, is a dominant negative regulator of transcription. J. Biol. Chem. 1996, 271, 13221–13227. [Google Scholar] [CrossRef] [PubMed]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms α and β have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. STATs: Transcriptional control and biological impact. Nat. Rev. Mol. Cell. Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kee, W.H.; Seow, K.T.; Fung, W.; Cao, X. The coiled-coil domain of STAT3 is essential for its SH2 domain-mediated receptor binding and subsequent activation induced by epidermal growth factor and interleukin-6. Mol. Cell. Biol. 2000, 20, 7132–7139. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Seidel, H.M.; Milocco, L.H.; Lamb, P.; Darnell, J.E., Jr.; Stein, R.B.; Rosen, J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proc. Natl. Acad. Sci. USA 1995, 92, 3041–3045. [Google Scholar] [CrossRef] [PubMed]
- Tkach, M.; Rosemblit, C.; Rivas, M.A.; Proietti, C.J.; Diaz Flaque, M.C.; Mercogliano, M.F.; Beguelin, W.; Maronna, E.; Guzman, P.; Gercovich, F.G.; et al. p42/p44 MAPK-mediated STAT3Ser727 phosphorylation is required for progestin-induced full activation of STAT3 and breast cancer growth. Endocr. Relat. Cancer 2013, 20, 197–212. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.P.; Cao, X. Structure, function, and regulation of STAT proteins. Mol. Biosyst. 2006, 2, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by STAT1 and STAT3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammineni, P.; Anugula, C.; Mohammed, F.; Anjaneyulu, M.; Larner, A.C.; Sepuri, N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013, 288, 4723–4732. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Ribeiro, M.; Bray, E.R.; Lee, D.H.; Yungher, B.J.; Mehta, S.T.; Thakor, K.A.; Diaz, F.; Lee, J.K.; Moraes, C.T.; et al. Enhanced Transcriptional Activity and Mitochondrial Localization of STAT3 Co-induce Axon Regrowth in the Adult Central Nervous System. Cell Rep. 2016, 15, 398–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial STAT3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.A.; Pawson, T. Phosphotyrosine signaling: Evolving a new cellular communication system. Cell 2010, 142, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dube, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases and the control of cellular signaling responses. Adv. Pharmacol. 1996, 36, 91–119. [Google Scholar] [PubMed]
- Wallace, M.J.; Fladd, C.; Batt, J.; Rotin, D. The second catalytic domain of protein tyrosine phosphatase δ (PTP δ) binds to and inhibits the first catalytic domain of PTP σ. Mol. Cell. Biol. 1998, 18, 2608–2616. [Google Scholar] [CrossRef] [PubMed]
- Pulido, R.; Serra-Pages, C.; Tang, M.; Streuli, M. The LAR/PTP δ/PTP σ subfamily of transmembrane protein-tyrosine-phosphatases: Multiple human LAR, PTP δ, and PTP σ isoforms are expressed in a tissue-specific manner and associate with the LAR-interacting protein LIP.1. Proc. Natl. Acad. Sci. USA 1995, 92, 11686–11690. [Google Scholar] [CrossRef] [PubMed]
- Funato, K.; Yamazumi, Y.; Oda, T.; Akiyama, T. Tyrosine phosphatase PTPRD suppresses colon cancer cell migration in coordination with CD44. Exp. Ther. Med. 2011, 2, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Brito, M.R.; Bixby, J.L. Differential activities in adhesion and neurite growth of fibronectin type III repeats in the PTP-δ extracellular domain. Int. J. Dev. Neurosci. 2006, 24, 425–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 9435–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallings, R.L.; Nair, P.; Maris, J.M.; Catchpoole, D.; McDermott, M.; O’Meara, A.; Breatnach, F. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 2006, 66, 3673–3680. [Google Scholar] [CrossRef] [PubMed]
- Purdie, K.J.; Lambert, S.R.; Teh, M.T.; Chaplin, T.; Molloy, G.; Raghavan, M.; Kelsell, D.P.; Leigh, I.M.; Harwood, C.A.; Proby, C.M.; et al. Allelic imbalances and microdeletions affecting the PTPRD gene in cutaneous squamous cell carcinomas detected using single nucleotide polymorphism microarray analysis. Genes Chromosomes Cancer 2007, 46, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Kim, J.S.; Cronin, J.C.; Sibenaller, Z.; Ryken, T.; Rosenberg, S.A.; Ressom, H.; Jean, W.; Bigner, D.; Yan, H.; et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008, 68, 10300–10306. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, B.; Fabius, A.W.; Wu, W.H.; Pedraza, A.; Brennan, C.W.; Schultz, N.; Pitter, K.L.; Bromberg, J.F.; Huse, J.T.; Holland, E.C.; et al. Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 8149–8154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyser, N.D.; Du, Y.; Li, H.; Lui, V.; Xiao, X.; Chan, T.A.; Grandis, J.R. Loss-of-Function PTPRD Mutations Lead to Increased STAT3 Activation and Sensitivity to STAT3 Inhibition in Head and Neck Cancer. PLoS ONE 2015, 10, e0135750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walia, V.; Prickett, T.D.; Kim, J.S.; Gartner, J.J.; Lin, J.C.; Zhou, M.; Rosenberg, S.A.; Elble, R.C.; Solomon, D.A.; Waldman, T.; et al. Mutational and functional analysis of the tumor-suppressor PTPRD in human melanoma. Hum. Mutat. 2014, 35, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Heguy, A. The protein tyrosine phosphatase receptor D, a broadly inactivated tumor suppressor regulating STAT function. Cell Cycle 2009, 8, 3063–3064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, A.; Wang, Z. Tumour suppressor function of protein tyrosine phosphatase receptor-T. Biosci. Rep. 2011, 31, 303–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Becka, S.; Craig, S.E.; Lodowski, D.T.; Brady-Kalnay, S.M.; Wang, Z. Cancer-derived mutations in the fibronectin III repeats of PTPRT/PTPρ inhibit cell-cell aggregation. Cell Commun. Adhes. 2009, 16, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Becka, S.; Zhang, P.; Craig, S.E.; Lodowski, D.T.; Wang, Z.; Brady-Kalnay, S.M. Characterization of the adhesive properties of the type IIb subfamily receptor protein tyrosine phosphatases. Cell Commun. Adhes. 2010, 17, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Becka, S.; Zhang, P.; Zhang, X.; Brady-Kalnay, S.M.; Wang, Z. Tumor-derived extracellular mutations of PTPRT/PTPρ are defective in cell adhesion. Mol. Cancer Res. 2008, 6, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Besco, J.A.; Hooft van Huijsduijnen, R.; Frostholm, A.; Rotter, A. Intracellular substrates of brain-enriched receptor protein tyrosine phosphatase ρ (RPTPρ/PTPRT). Brain Res. 2006, 1116, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Kwon, S.K.; Lee, M.K.; Moon, J.; Jeong, D.G.; Park, E.; Kim, S.J.; Park, B.C.; Lee, S.C.; Ryu, S.E.; et al. Synapse formation regulated by protein tyrosine phosphatase receptor T through interaction with cell adhesion molecules and Fyn. EMBO J. 2009, 28, 3564–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Zhang, X.; Guda, K.; Lawrence, E.; Sun, Q.; Watanabe, T.; Iwakura, Y.; Asano, M.; Wei, L.; Yang, Z.; et al. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc. Natl. Acad. Sci. USA 2010, 107, 2592–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Guo, A.; Yu, J.; Possemato, A.; Chen, Y.; Zheng, W.; Polakiewicz, R.D.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E.; et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc. Natl. Acad. Sci. USA 2007, 104, 4060–4064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Shen, D.; Parsons, D.W.; Bardelli, A.; Sager, J.; Szabo, S.; Ptak, J.; Silliman, N.; Peters, B.A.; van der Heijden, M.S.; et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 2004, 304, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Peyser, N.D.; Ng, P.K.; Hritz, J.; Zeng, Y.; Lu, Y.; Li, H.; Wang, L.; Gilbert, B.R.; General, I.J.; et al. Frequent mutation of receptor protein tyrosine phosphatases provides a mechanism for STAT3 hyperactivation in head and neck cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1114–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Scott, A.; Zhang, P.; Hao, Y.; Feng, X.; Somasundaram, S.; Khalil, A.M.; Willis, J.; Wang, Z. Regulation of paxillin-p130-PI3K-AKT signaling axis by Src and PTPRT impacts colon tumorigenesis. Oncotarget 2017, 8, 48782–48793. [Google Scholar] [CrossRef] [PubMed]
- Peyser, N.D.; Freilino, M.; Wang, L.; Zeng, Y.; Li, H.; Johnson, D.E.; Grandis, J.R. Frequent promoter hypermethylation of PTPRT increases STAT3 activation and sensitivity to STAT3 inhibition in head and neck cancer. Oncogene 2016, 35, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Li, N.; Fan, K.; Yin, P.; Zhao, C.; Li, Z.; Lin, Y.; Wang, L.; Zha, X. β1,6 GlcNAc branches-modified PTPRT attenuates its activity and promotes cell migration by STAT3 pathway. PLoS ONE 2014, 9, e98052. [Google Scholar] [CrossRef] [PubMed]
- Sap, J.; Jiang, Y.P.; Friedlander, D.; Grumet, M.; Schlessinger, J. Receptor tyrosine phosphatase R-PTP-κ mediates homophilic binding. Mol. Cell. Biol. 1994, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Shin, I.; Qu, S.; Arteaga, C.L. Transforming growth factor β (TGF-β)-Smad target gene protein tyrosine phosphatase receptor type κ is required for TGF-β function. Mol. Cell. Biol. 2005, 25, 4703–4715. [Google Scholar] [CrossRef] [PubMed]
- Drosopoulos, N.E.; Walsh, F.S.; Doherty, P. A soluble version of the receptor-like protein tyrosine phosphatase κ stimulates neurite outgrowth via a Grb2/MEK1-dependent signaling cascade. Mol. Cell. Neurosci. 1999, 13, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Kose, H.; Sakai, T.; Tsukumo, S.; Wei, K.; Yamada, T.; Yasutomo, K.; Matsumoto, K. Maturational arrest of thymocyte development is caused by a deletion in the receptor-like protein tyrosine phosphatase κ gene in LEC rats. Genomics 2007, 89, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Schnekenburger, J.; Mayerle, J.; Kruger, B.; Buchwalow, I.; Weiss, F.U.; Albrecht, E.; Samoilova, V.E.; Domschke, W.; Lerch, M.M. Protein tyrosine phosphatase κ and SHP-1 are involved in the regulation of cell-cell contacts at adherens junctions in the exocrine pancreas. Gut 2005, 54, 1445–1455. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Muller, T.; Lerch, M.M.; Ullrich, A. Association of human protein-tyrosine phosphatase κ with members of the armadillo family. J. Biol. Chem. 1996, 271, 16712–16719. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.H.; Ye, L.; Mason, M.D.; Jiang, W.G. Protein tyrosine phosphatase κ (PTPRK) is a negative regulator of adhesion and invasion of breast cancer cells, and associates with poor prognosis of breast cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Kishi, M.; Sakaki, T.; Hashimoto, H.; Nakase, H.; Shimada, K.; Ishida, E.; Konishi, N. Novel tumor suppressor loci on 6q22-23 in primary central nervous system lymphomas. Cancer Res. 2003, 63, 737–741. [Google Scholar] [PubMed]
- Starr, T.K.; Allaei, R.; Silverstein, K.A.; Staggs, R.A.; Sarver, A.L.; Bergemann, T.L.; Gupta, M.; O’Sullivan, M.G.; Matise, I.; Dupuy, A.J.; et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science 2009, 323, 1747–1750. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.H.; Ye, L.; Mason, M.D.; Jiang, W.G. Receptor-like protein tyrosine phosphatase κ negatively regulates the apoptosis of prostate cancer cells via the JNK pathway. Int. J. Oncol. 2013, 43, 1560–1568. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Al-Keilani, M.S.; Alqudah, M.A.; Sibenaller, Z.A.; Ryken, T.C.; Assem, M. Tumor derived mutations of protein tyrosine phosphatase receptor type K affect its function and alter sensitivity to chemotherapeutics in glioma. PLoS ONE 2013, 8, e62852. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tan, L.J.; Grachtchouk, V.; Voorhees, J.J.; Fisher, G.J. Receptor-type protein-tyrosine phosphatase-κ regulates epidermal growth factor receptor function. J. Biol. Chem. 2005, 280, 42694–42700. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, Y.; Yang, Z.; Liu, M.; Li, Z.; Sun, L.; Mei, C.; Chen, H.; Chen, L.; Wang, L.; et al. EGF-mediated migration signaling activated by N-acetylglucosaminyltransferase-V via receptor protein tyrosine phosphatase κ. Arch. Biochem. Biophys. 2009, 486, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xue, S.; Zhou, J.; Voorhees, J.J.; Fisher, G.J. Notch and TGF-β pathways cooperatively regulate receptor protein tyrosine phosphatase-κ (PTPRK) gene expression in human primary keratinocytes. Mol. Biol. Cell 2015, 26, 1199–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.W.; Guo, T.; Shen, L.; Wong, K.Y.; Tao, Q.; Choi, W.W.; Au-Yeung, R.K.; Chan, Y.P.; Wong, M.L.; Tang, J.C.; et al. Receptor-type tyrosine-protein phosphatase κ directly targets STAT3 activation for tumor suppression in nasal NK/T-cell lymphoma. Blood 2015, 125, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Somani, A.K.; Siminovitch, K.A. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin. Immunol. 2000, 12, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Wang, W.; Liu, L.; Song, X.; Mo, Y.; Komma, C.; Bellamy, H.D.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein tyrosine phosphatase SHP-1 in the open conformation. J. Cell. Biochem. 2011, 112, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Sun, M.; Liu, L.; Zhou, G.W. The function of the protein tyrosine phosphatase SHP-1 in cancer. Gene 2003, 306, 1–12. [Google Scholar] [CrossRef]
- Yang, J.; Liu, L.; He, D.; Song, X.; Liang, X.; Zhao, Z.J.; Zhou, G.W. Crystal structure of human protein-tyrosine phosphatase SHP-1. J. Biol. Chem. 2003, 278, 6516–6520. [Google Scholar] [CrossRef] [PubMed]
- Frank, C.; Burkhardt, C.; Imhof, D.; Ringel, J.; Zschornig, O.; Wieligmann, K.; Zacharias, M.; Bohmer, F.D. Effective dephosphorylation of Src substrates by SHP-1. J. Biol. Chem. 2004, 279, 11375–11383. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Amin, H.M.; Franko, B.; Frantz, C.; Shi, X.; Lai, R. Loss of SHP1 enhances JAK3/STAT3 signaling and decreases proteosome degradation of JAK3 and NPM-ALK in ALK+ anaplastic large-cell lymphoma. Blood 2006, 108, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Rassidakis, G.Z.; Medeiros, L.J.; Amin, H.M.; Lai, R. Methylation of SHP1 gene and loss of SHP1 protein expression are frequent in systemic anaplastic large cell lymphoma. Blood 2004, 104, 1580–1581. [Google Scholar] [CrossRef] [PubMed]
- Chim, C.S.; Fung, T.K.; Cheung, W.C.; Liang, R.; Kwong, Y.L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the JAK/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Ouchida, M.; Koyama, M.; Ogama, Y.; Takada, S.; Nakatani, Y.; Tanaka, T.; Yoshino, T.; Hayashi, K.; Ohara, N.; et al. Gene silencing of the tyrosine phosphatase SHP1 gene by aberrant methylation in leukemias/lymphomas. Cancer Res. 2002, 62, 6390–6394. [Google Scholar] [PubMed]
- Koyama, M.; Oka, T.; Ouchida, M.; Nakatani, Y.; Nishiuchi, R.; Yoshino, T.; Hayashi, K.; Akagi, T.; Seino, Y. Activated proliferation of B-cell lymphomas/leukemias with the SHP1 gene silencing by aberrant CpG methylation. Lab. Investig. 2003, 83, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.; Raghunath, P.; Wasik, A.; Junkins-Hopkins, J.M.; Jones, D.; Zhang, Q.; Odum, N.; Wasik, M.A. Loss of SHP-1 tyrosine phosphatase expression correlates with the advanced stages of cutaneous T-cell lymphoma. Hum. Pathol. 2007, 38, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, H.Y.; Marzec, M.; Raghunath, P.N.; Nagasawa, T.; Wasik, M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6948–6953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demosthenous, C.; Han, J.J.; Hu, G.; Stenson, M.; Gupta, M. Loss of function mutations in PTPN6 promote STAT3 deregulation via JAK3 kinase in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 44703–44713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, K.S.; Sethi, G.; Sung, B.; Goel, A.; Ralhan, R.; Aggarwal, B.B. Guggulsterone, a farnesoid X receptor antagonist, inhibits constitutive and inducible STAT3 activation through induction of a protein tyrosine phosphatase SHP-1. Cancer Res. 2008, 68, 4406–4415. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Cheng, A.L.; Shiau, C.W.; Liu, C.Y.; Ko, C.H.; Lin, M.W.; Chen, P.J.; Chen, K.F. Dovitinib induces apoptosis and overcomes sorafenib resistance in hepatocellular carcinoma through SHP-1-mediated inhibition of STAT3. Mol. Cancer Ther. 2012, 11, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.F.; Tai, W.T.; Hsu, C.Y.; Huang, J.W.; Liu, C.Y.; Chen, P.J.; Kim, I.; Shiau, C.W. Blockade of STAT3 activation by sorafenib derivatives through enhancing SHP-1 phosphatase activity. Eur. J. Med. Chem. 2012, 55, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Sandur, S.K.; Pandey, M.K.; Sung, B.; Aggarwal, B.B. 5-hydroxy-2-methyl-1,4-naphthoquinone, a vitamin K3 analogue, suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase, SHP-1: Potential role in chemosensitization. Mol. Cancer Res. 2010, 8, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Tremblay, M.L.; Digiovanni, J. Protein tyrosine phosphatases, TC-PTP, SHP1, and SHP2, cooperate in rapid dephosphorylation of STAT3 in keratinocytes following UVB irradiation. PLoS ONE 2010, 5, e10290. [Google Scholar] [CrossRef] [PubMed]
- Ruchusatsawat, K.; Wongpiyabovorn, J.; Shuangshoti, S.; Hirankarn, N.; Mutirangura, A. SHP-1 promoter 2 methylation in normal epithelial tissues and demethylation in psoriasis. J. Mol. Med. (Berl.) 2006, 84, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Lewis, R.S.; Babon, J.J.; Mintern, J.D.; Jenne, D.E.; Metcalf, D.; Zhang, J.G.; Cengia, L.H.; O’Donnell, J.A.; Roberts, A.W. Neutrophils require SHP1 to regulate IL-1β production and prevent inflammatory skin disease. J. Immunol. 2011, 186, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Modi, H.; McDonald, T.; Rossi, J.; Yee, J.K.; Bhatia, R. A critical role for SHP2 in STAT5 activation and growth factor-mediated proliferation, survival, and differentiation of human CD34+ cells. Blood 2011, 118, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Hong, H.; Kawakami, Y.; Kato, Y.; Wu, D.; Yasudo, H.; Kimura, A.; Kubagawa, H.; Bertoli, L.F.; Davis, R.S.; et al. Tumor suppression by phospholipase C-β3 via SHP-1-mediated dephosphorylation of STAT5. Cancer Cell 2009, 16, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Xiao, W.; Gao, P.; Namiranian, S.; Matsumoto, K.; Tomimori, Y.; Hong, H.; Yamashita, H.; Kimura, M.; Kashiwakura, J.; et al. Critical role for mast cell STAT5 activity in skin inflammation. Cell Rep. 2014, 6, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.M.; Dickensheets, H.; Qu, C.K.; Donnelly, R.P.; Keegan, A.D. Regulation of the dephosphorylation of STAT6. Participation of Tyr-713 in the interleukin-4 receptor α, the tyrosine phosphatase SHP-1, and the proteasome. J. Biol. Chem. 2003, 278, 3903–3911. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Coleman, J.M.; Su, Y.; Mann, M.; Ryan, J.; Shultz, L.D.; Huang, H. SHP-1 regulates STAT6 phosphorylation and IL-4-mediated function in a cell type-specific manner. Cytokine 2005, 29, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.J.; Pao, L.I.; Dhanji, S.; Murakami, K.; Ohashi, P.S.; Neel, B.G. Shp1 regulates T cell homeostasis by limiting IL-4 signals. J. Exp. Med. 2013, 210, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.H.; Qu, C.K.; Henegariu, O.; Lu, X.; Feng, G.S. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J. Biol. Chem. 1998, 273, 21125–21131. [Google Scholar] [CrossRef] [PubMed]
- Bentires-Alj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.; Bardelli, A.; Sugarbaker, D.J.; et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed]
- Mohi, M.G.; Neel, B.G. The role of SHP2 (PTPN11) in cancer. Curr. Opin. Genet. Dev. 2007, 17, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Hellberg, C.; Bohmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Ivins Zito, C.; Kontaridis, M.I.; Fornaro, M.; Feng, G.S.; Bennett, A.M. SHP-2 regulates the phosphatidylinositide 3’-kinase/Akt pathway and suppresses caspase 3-mediated apoptosis. J. Cell. Physiol. 2004, 199, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Kontaridis, M.I.; Swanson, K.D.; David, F.S.; Barford, D.; Neel, B.G. PTPN11 (SHP2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 2006, 281, 6785–6792. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.J.; Feng, G.S. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2007, 109, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Niu, R. Functions of SHP2 in cancer. J. Cell. Mol. Med. 2015, 19, 2075–2083. [Google Scholar] [CrossRef] [PubMed]
- Bard-Chapeau, E.A.; Li, S.; Ding, J.; Zhang, S.S.; Zhu, H.H.; Princen, F.; Fang, D.D.; Han, T.; Bailly-Maitre, B.; Poli, V.; et al. PTPN11/SHP2 acts as a tumor suppressor in hepatocellular carcinogenesis. Cancer Cell 2011, 19, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Han, T.; Tang, H.; Huang, K.; Min, J.; Li, J.; Ding, X.; Xu, Z. SHP2 Inhibits Proliferation of Esophageal Squamous Cell Cancer via Dephosphorylation of STAT3. Int. J. Mol. Sci. 2017, 18, 134. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.; Cao, F.; Jiang, H.; Li, A.; Li, J.; Qiu, L.; Shen, H.; Chang, W.; Zhou, C.; et al. SHP2 associates with nuclear localization of STAT3: Significance in progression and prognosis of colorectal cancer. Sci. Rep. 2017, 7, 17597. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Kuang, Y.; Zhang, C.; Zhang, Z.; Chen, L.; Li, B.; Li, Y.; Wang, Y.; Yang, H.; Han, Q.; et al. Glucose-6-phosphate dehydrogenase and NADPH oxidase 4 control STAT3 activity in melanoma cells through a pathway involving reactive oxygen species, c-SRC and SHP2. Am. J. Cancer Res. 2015, 5, 1610–1620. [Google Scholar] [PubMed]
- Ramana, C.V.; Chatterjee-Kishore, M.; Nguyen, H.; Stark, G.R. Complex roles of STAT1 in regulating gene expression. Oncogene 2000, 19, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.R.; Hong, Y.K.; Wang, X.D.; Ling, M.Y.; Dragoi, A.M.; Chung, A.S.; Campbell, A.G.; Han, Z.Y.; Feng, G.S.; Chin, Y.E. SHP-2 is a dual-specificity phosphatase involved in STAT1 dephosphorylation at both tyrosine and serine residues in nuclei. J. Biol. Chem. 2002, 277, 47572–47580. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhao, Y.; Fang, J.; Cui, R.; Xiao, Y.; Xu, Q. SHP2 negatively regulates HLA-ABC and PD-L1 expression via STAT1 phosphorylation in prostate cancer cells. Oncotarget 2017, 8, 53518–53530. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.S.; Srivastava, R.M.; Andrade Filho, P.A.; Egloff, A.M.; Wang, L.; Seethala, R.R.; Ferrone, S.; Ferris, R.L. SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T-cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin. Cancer Res. 2013, 19, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wen, R.; Yang, S.; Schuman, J.; Zhang, E.E.; Yi, T.; Feng, G.S.; Wang, D. Identification of SHP-2 as a STAT5A phosphatase. J. Biol. Chem. 2003, 278, 16520–16527. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Samten, B.; Ji, H.L.; Zhao, Z.J.; Tang, H. Tyrosine phosphatase PTP-MEG2 negatively regulates vascular endothelial growth factor receptor signaling and function in endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C548–C553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Marlin, M.C.; Liang, Z.; Ahmad, M.; Ashpole, N.M.; Sonntag, W.E.; Zhao, Z.J.; Li, G. The Protein Tyrosine Phosphatase MEG2 Regulates the Transport and Signal Transduction of Tropomyosin Receptor Kinase A. J. Biol. Chem. 2016, 291, 23895–23905. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Warshawsky, I.; Majerus, P.W. Cloning and expression of a cytosolic megakaryocyte protein-tyrosine-phosphatase with sequence homology to retinaldehyde-binding protein and yeast SEC14p. Proc. Natl. Acad. Sci. USA 1992, 89, 2980–2984. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Williams, S.; Bulankina, A.; Honing, S.; Mustelin, T. Association of protein-tyrosine phosphatase MEG2 via its Sec14p homology domain with vesicle-trafficking proteins. J. Biol. Chem. 2007, 282, 15170–15178. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Sui, X.; Zhao, R.; Dai, C.; Krantz, S.B.; Zhao, Z.J. PTP-MEG2 is activated in polycythemia vera erythroid progenitor cells and is required for growth and expansion of erythroid cells. Blood 2003, 102, 4354–4360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Vachon, E.; Zhang, J.; Cherepanov, V.; Kruger, J.; Li, J.; Saito, K.; Shannon, P.; Bottini, N.; Huynh, H.; et al. Tyrosine phosphatase MEG2 modulates murine development and platelet and lymphocyte activation through secretory vesicle function. J. Exp. Med. 2005, 202, 1587–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, C.Y.; Koo, S.H.; Wang, Y.; Callaway, S.; Hedrick, S.; Mak, P.A.; Orth, A.P.; Peters, E.C.; Saez, E.; Montminy, M.; et al. Identification of the tyrosine phosphatase PTP-MEG2 as an antagonist of hepatic insulin signaling. Cell Metab. 2006, 3, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, F.; Ren, F.; Rong, Y.; Wang, Y.; Geng, Y.; Wang, Y.; Feng, M.; Ju, Y.; Li, Y.; Zhao, Z.J.; et al. Protein tyrosine phosphatase Meg2 dephosphorylates signal transducer and activator of transcription 3 and suppresses tumor growth in breast cancer. Breast Cancer Res. 2012, 14, R38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Kim, Y.H.; Park, J.Y.; Lee, Y.J.; Oh, H.M.; Choi, S.K.; Han, D.C.; Kwon, B.M. Methyllucidone inhibits STAT3 activity by regulating the expression of the protein tyrosine phosphatase MEG2 in DU145 prostate carcinoma cells. Bioorg Med. Chem. Lett. 2018, 28, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Barford, D.; Flint, A.J.; Tonks, N.K. Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 1995, 268, 1754–1758. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, A.; Dube, N.; Tremblay, M.L. Cytoplasmic protein tyrosine phosphatases, regulation and function: The roles of PTP1B and TC-PTP. Curr. Opin. Cell Biol. 2005, 17, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Tillmann, U.; Wagner, J.; Boerboom, D.; Westphal, H.; Tremblay, M.L. Nuclear localization and cell cycle regulation of a murine protein tyrosine phosphatase. Mol. Cell. Biol. 1994, 14, 3030–3040. [Google Scholar] [CrossRef] [PubMed]
- Kamatkar, S.; Radha, V.; Nambirajan, S.; Reddy, R.S.; Swarup, G. Two splice variants of a tyrosine phosphatase differ in substrate specificity, DNA binding, and subcellular location. J. Biol. Chem. 1996, 271, 26755–26761. [Google Scholar] [CrossRef] [PubMed]
- Cool, D.E.; Tonks, N.K.; Charbonneau, H.; Walsh, K.A.; Fischer, E.H.; Krebs, E.G. cDNA isolated from a human T-cell library encodes a member of the protein-tyrosine-phosphatase family. Proc. Natl. Acad. Sci. USA 1989, 86, 5257–5261. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Klingler-Hoffmann, M.; Fodero-Tavoletti, M.T.; Puryer, M.A.; Meng, T.C.; Tonks, N.K.; Tiganis, T. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol. Cell. Biol. 2003, 23, 2096–2108. [Google Scholar] [CrossRef] [PubMed]
- Simoncic, P.D.; McGlade, C.J.; Tremblay, M.L. PTP1B and TC-PTP: Novel roles in immune-cell signaling. Can. J. Physiol. Pharmacol. 2006, 84, 667–675. [Google Scholar] [CrossRef] [PubMed]
- You-Ten, K.E.; Muise, E.S.; Itie, A.; Michaliszyn, E.; Wagner, J.; Jothy, S.; Lapp, W.S.; Tremblay, M.L. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J. Exp. Med. 1997, 186, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, K.M.; Nestel, F.P.; Newell, E.W.; Charette, G.; Seemayer, T.A.; Tremblay, M.L.; Lapp, W.S. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood 2004, 103, 3457–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, K.A.; Hatzihristidis, T.; Bussieres-Marmen, S.; Robert, F.; Desai, N.; Miranda-Saavedra, D.; Pelletier, J.; Tremblay, M.L. TC-PTP regulates the IL-7 transcriptional response during murine early T cell development. Sci. Rep. 2017, 7, 13275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiganis, T.; Kemp, B.E.; Tonks, N.K. The protein-tyrosine phosphatase TCPTP regulates epidermal growth factor receptor-mediated and phosphatidylinositol 3-kinase-dependent signaling. J. Biol. Chem. 1999, 274, 27768–27775. [Google Scholar] [CrossRef] [PubMed]
- Simoncic, P.D.; Lee-Loy, A.; Barber, D.L.; Tremblay, M.L.; McGlade, C.J. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr. Biol. 2002, 12, 446–453. [Google Scholar] [CrossRef]
- Fukushima, A.; Loh, K.; Galic, S.; Fam, B.; Shields, B.; Wiede, F.; Tremblay, M.L.; Watt, M.J.; Andrikopoulos, S.; Tiganis, T. T-cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes 2010, 59, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Kleppe, M.; Lahortiga, I.; El Chaar, T.; De Keersmaecker, K.; Mentens, N.; Graux, C.; Van Roosbroeck, K.; Ferrando, A.A.; Langerak, A.W.; Meijerink, J.P.; et al. Deletion of the protein tyrosine phosphatase gene PTPN2 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 530–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleppe, M.; Tousseyn, T.; Geissinger, E.; Kalender Atak, Z.; Aerts, S.; Rosenwald, A.; Wlodarska, I.; Cools, J. Mutation analysis of the tyrosine phosphatase PTPN2 in Hodgkin’s lymphoma and T-cell non-Hodgkin’s lymphoma. Haematologica 2011, 96, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.J.; Wiede, F.; Gurzov, E.N.; Wee, K.; Hauser, C.; Zhu, H.J.; Molloy, T.J.; O’Toole, S.A.; Daly, R.J.; Sutherland, R.L.; et al. TCPTP regulates SFK and STAT3 signaling and is lost in triple-negative breast cancers. Mol. Cell. Biol. 2013, 33, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Sano, S.; Kataoka, K.; Abel, E.; Carbajal, S.; Beltran, L.; Clifford, J.; Peavey, M.; Shen, J.; Digiovanni, J. Forced expression of a constitutively active form of STAT3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene 2008, 27, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Sano, S.; Kiguchi, K.; Anders, J.; Komazawa, N.; Takeda, J.; DiGiovanni, J. Disruption of STAT3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J. Clin. Investig. 2004, 114, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Chan, K.S.; Sano, S.; Digiovanni, J. Signal transducer and activator of transcription 3 (STAT3) in epithelial carcinogenesis. Mol. Carcinog 2007, 46, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Kataoka, K.; Rao, D.; Kiguchi, K.; Cotsarelis, G.; Digiovanni, J. Targeted disruption of stat3 reveals a major role for follicular stem cells in skin tumor initiation. Cancer Res. 2009, 69, 7587–7594. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Angel, J.M.; Sano, S.; DiGiovanni, J. Constitutive activation and targeted disruption of signal transducer and activator of transcription 3 (STAT3) in mouse epidermis reveal its critical role in UVB-induced skin carcinogenesis. Oncogene 2009, 28, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Kim, D.J.; Carbajal, S.; Clifford, J.L.; DiGiovanni, J. Stage-specific disruption of STAT3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis 2008, 29, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Baek, M.; Kim, D.J. Protein Tyrosine Signaling and its Potential Therapeutic Implications in Carcinogenesis. Curr. Pharm. Des. 2017, 23, 4226–4246. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Morales, L.D.; Slaga, T.J.; Kim, D.J. Activation of T-cell protein-tyrosine phosphatase suppresses keratinocyte survival and proliferation following UVB irradiation. J. Biol. Chem. 2015, 290, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Morales, L.D.; Baek, M.; Slaga, T.J.; DiGiovanni, J.; Kim, D.J. UVB-induced nuclear translocation of TC-PTP by AKT/14-3-3σ axis inhibits keratinocyte survival and proliferation. Oncotarget 2017, 8, 90674–90692. [Google Scholar] [PubMed]
- Kim, M.; Morales, L.D.; Kim, D.J. TC-PTP nuclear trafficking in keratinocytes. Aging (Albany NY) 2017, 9, 2459–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Kim, M.; Baek, M.; Morales, L.D.; Jang, I.S.; Slaga, T.J.; DiGiovanni, J.; Kim, D.J. Targeted disruption of TC-PTP in the proliferative compartment augments STAT3 and AKT signaling and skin tumor development. Sci. Rep. 2017, 7, 45077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, W.; Zhang, L.; Yang, G.; Zhai, J.; Hu, Z.; Chen, Y.; Chen, X.; Hui, L.; Huang, R.; Hu, G. Nuclear β-arrestin1 functions as a scaffold for the dephosphorylation of STAT1 and moderates the antiviral activity of IFN-γ. Mol. Cell 2008, 31, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Pelzel, C.; Begitt, A.; Wenta, N.; Vinkemeier, U. Evidence against a role for β-arrestin1 in STAT1 dephosphorylation and the inhibition of interferon-γ signaling. Mol. Cell 2013, 50, 149–156. [Google Scholar] [CrossRef] [PubMed]
- ten Hoeve, J.; de Jesus Ibarra-Sanchez, M.; Fu, Y.; Zhu, W.; Tremblay, M.; David, M.; Shuai, K. Identification of a nuclear STAT1 protein tyrosine phosphatase. Mol. Cell. Biol. 2002, 22, 5662–5668. [Google Scholar] [CrossRef] [PubMed]
- Bussieres-Marmen, S.; Vinette, V.; Gungabeesoon, J.; Aubry, I.; Perez-Quintero, L.A.; Tremblay, M.L. Loss of T-cell protein tyrosine phosphatase in the intestinal epithelium promotes local inflammation by increasing colonic stem cell proliferation. Cell. Mol. Immunol. 2018, 15, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Festen, E.A.; Goyette, P.; Green, T.; Boucher, G.; Beauchamp, C.; Trynka, G.; Dubois, P.C.; Lagace, C.; Stokkers, P.C.; Hommes, D.W.; et al. A meta-analysis of genome-wide association scans identifies IL18RAP, PTPN2, TAGAP, and PUS10 as shared risk loci for Crohn’s disease and celiac disease. PLoS Genet. 2011, 7, e1001283. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.; Morales, L.D.; Jang, I.-S.; Cho, Y.-Y.; Kim, D.J. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. Int. J. Mol. Sci. 2018, 19, 2708. https://doi.org/10.3390/ijms19092708
Kim M, Morales LD, Jang I-S, Cho Y-Y, Kim DJ. Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. International Journal of Molecular Sciences. 2018; 19(9):2708. https://doi.org/10.3390/ijms19092708
Chicago/Turabian StyleKim, Mihwa, Liza D. Morales, Ik-Soon Jang, Yong-Yeon Cho, and Dae Joon Kim. 2018. "Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling" International Journal of Molecular Sciences 19, no. 9: 2708. https://doi.org/10.3390/ijms19092708
APA StyleKim, M., Morales, L. D., Jang, I. -S., Cho, Y. -Y., & Kim, D. J. (2018). Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling. International Journal of Molecular Sciences, 19(9), 2708. https://doi.org/10.3390/ijms19092708