The Pathways of the iRFP713 Unfolding Induced by Different Denaturants

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
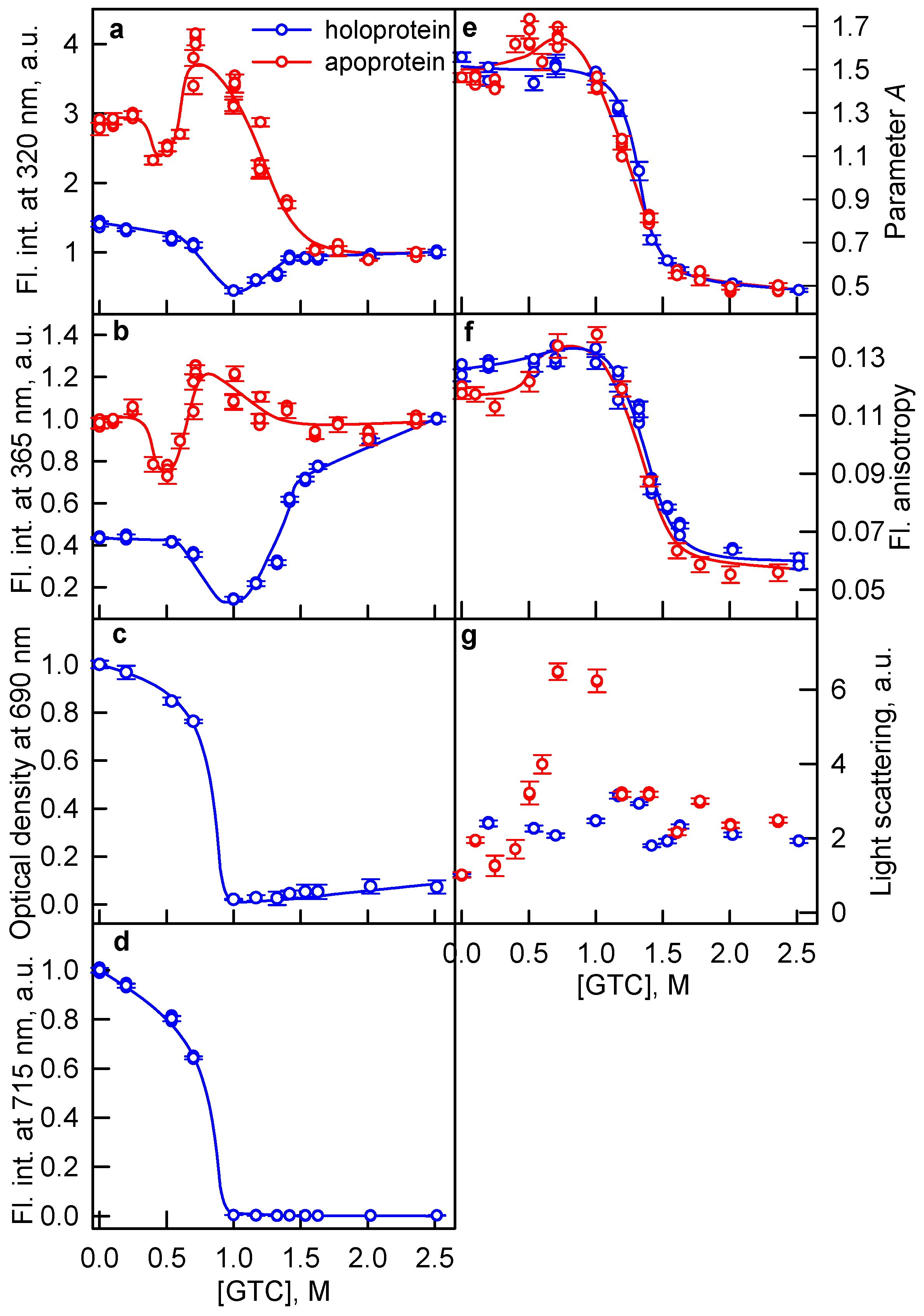
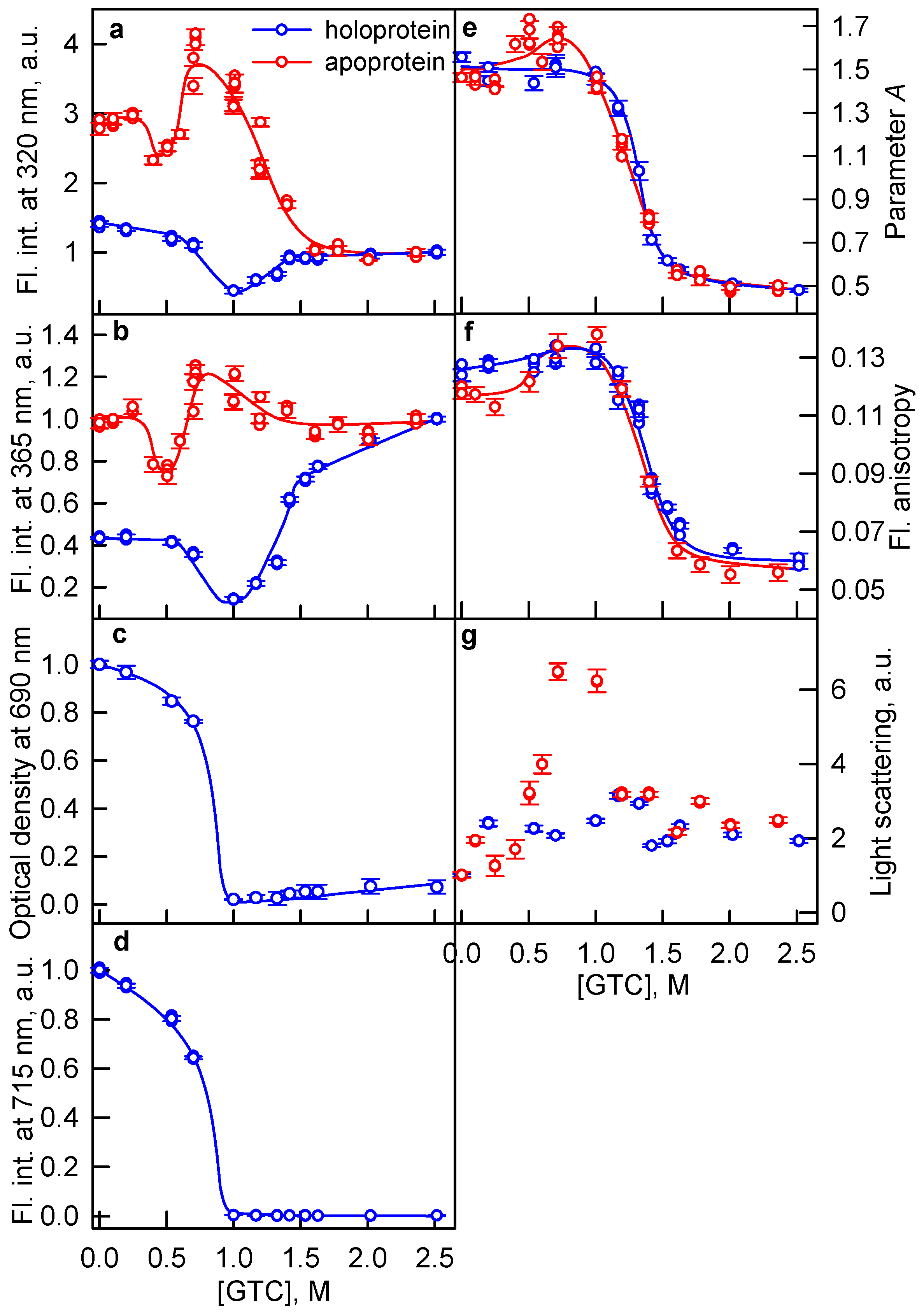
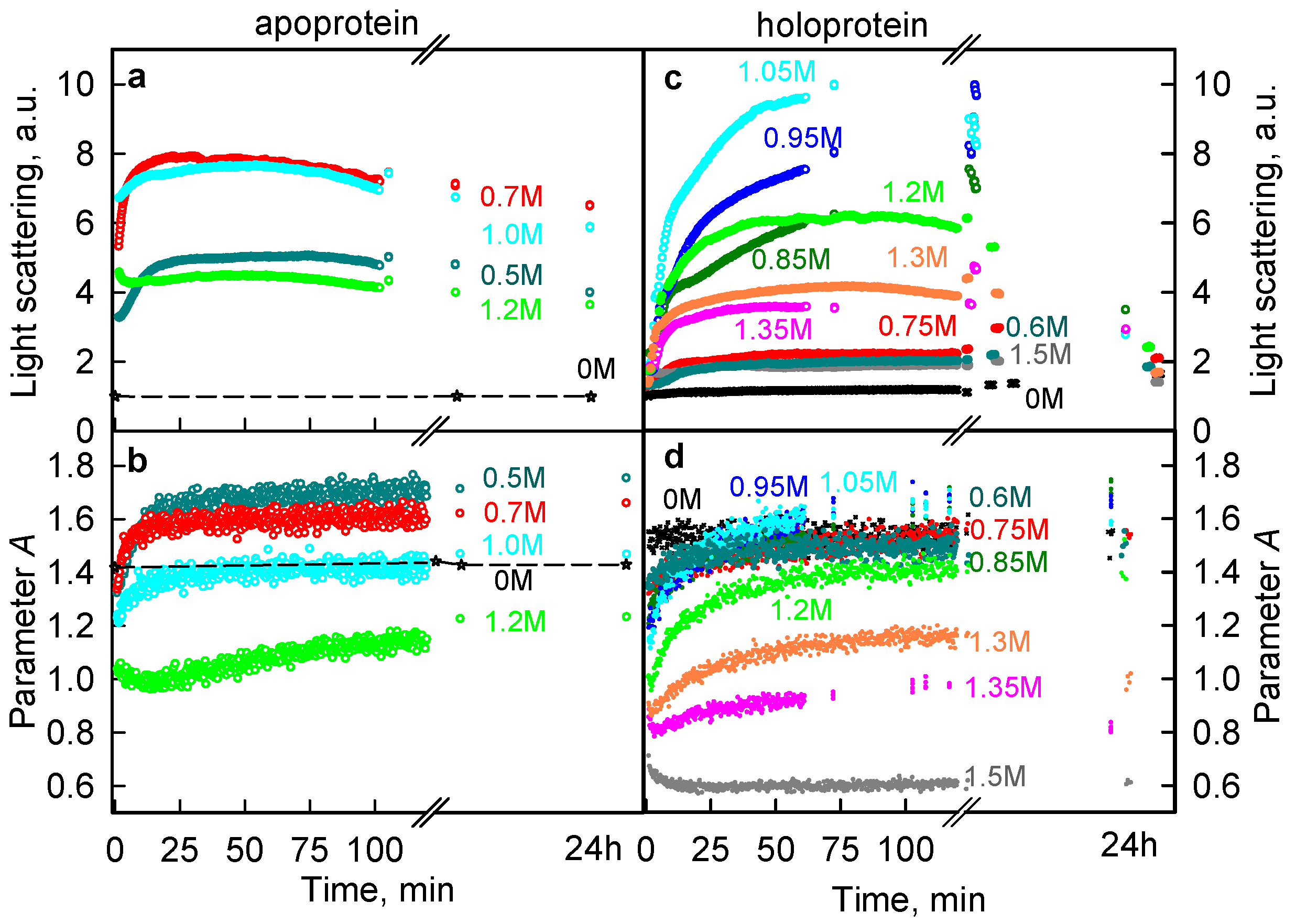
2.1. The Unfolding of iRFP713 Is Accompanied by the Protein Aggregation
2.2. Formation of the Monomeric State at Unfolding of iRFP713 in the Holoform
2.3. Reasons of the Aggregation of iRFP713 in the Apo- and Holoform
3. Materials and Methods
3.1. Plasmids, Mutagenesis, Protein Expression, and Purification
3.2. Spectrophotometric Experiments
3.3. Fluorescence Spectroscopy
3.4. Gel Filtration Experiments
3.5. Circular Dichroism Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chernov, K.G.; Redchuk, T.A.; Omelina, E.S.; Verkhusha, V.V. Near-Infrared Fluorescent Proteins, Biosensors, and Optogenetic Tools Engineered from Phytochromes. Chem. Rev. 2017, 117, 6423–6446. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, E.A.; Campbell, R.E.; Lin, J.Y.; Lin, M.Z.; Miyawaki, A.; Palmer, A.E.; Shu, X.; Zhang, J.; Tsien, R.Y. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci. 2017, 42, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rockwell, N.C.; Lagarias, J.C. A brief history of phytochromes. Chemphyschem 2010, 11, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Ulijasz, A.T.; Vierstra, R.D. Phytochrome structure and photochemistry: Recent advances toward a complete molecular picture. Curr. Opin. Plant Biol. 2011, 14, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Bhoo, S.H.; Davis, S.J.; Walker, J.; Karniol, B.; Vierstra, R.D. Bacteriophytochromes are photochromic histidine kinases using a biliverdin chromophore. Nature 2001, 414, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Kapitulnik, J.; Maines, M.D. The role of bile pigments in health and disease: Effects on cell signaling, cytotoxicity, and cytoprotection. Front. Pharmacol. 2012, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, O.V.; Baloban, M.; Bublikov, G.S.; Shcherbakova, D.M.; Stepanenko, O.V.; Turoverov, K.K.; Kuznetsova, I.M.; Verkhusha, V.V. Allosteric effects of chromophore interaction with dimeric near-infrared fluorescent proteins engineered from bacterial phytochromes. Sci. Rep. 2016, 6, 18750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shcherbakova, D.M.; Baloban, M.; Pletnev, S.; Malashkevich, V.N.; Xiao, H.; Dauter, Z.; Verkhusha, V.V. Molecular Basis of Spectral Diversity in Near-Infrared Phytochrome-Based Fluorescent Proteins. Chem. Biol. 2015, 22, 1540–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanenko, O.V.; Stepanenko, O.V.; Bublikov, G.S.; Kuznetsova, I.M.; Verkhusha, V.V.; Turoverov, K.K. Stabilization of structure in near-infrared fluorescent proteins by binding of biliverdin chromophore. J. Mol. Struct. 2017, 1140, 22–31. [Google Scholar] [CrossRef]
- Stepanenko, O.V.; Stepanenko, O.V.; Kuznetsova, I.M.; Shcherbakova, D.M.; Verkhusha, V.V.; Turoverov, K.K. Interaction of Biliverdin Chromophore with Near-Infrared Fluorescent Protein BphP1-FP Engineered from Bacterial Phytochrome. Int. J. Mol. Sci. 2017, 18, 1009. [Google Scholar] [CrossRef] [PubMed]
- Filonov, G.S.; Piatkevich, K.D.; Ting, L.M.; Zhang, J.; Kim, K.; Verkhusha, V.V. Bright and stable near-infrared fluorescent protein for in vivo imaging. Nat. Biotechnol. 2011, 29, 757–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanenko, O.V.; Bublikov, G.S.; Stepanenko, O.V.; Shcherbakova, D.M.; Verkhusha, V.V.; Turoverov, K.K.; Kuznetsova, I.M. A knot in the protein structure-probing the near-infrared fluorescent protein iRFP designed from a bacterial phytochrome. FEBS J. 2014, 281, 2284–2298. [Google Scholar] [CrossRef] [PubMed]
- Jamroz, M.; Niemyska, W.; Rawdon, E.J.; Stasiak, A.; Millett, K.C.; Sulkowski, P.; Sulkowska, J.I. KnotProt: A database of proteins with knots and slipknots. Nucleic Acids Res. 2015, 43, D306–D314. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Zhang, J.; Brunzelle, J.S.; Vierstra, R.D.; Forest, K.T. High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J. Biol. Chem. 2007, 282, 12298–12309. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Yamada, T.; Carlsson, U.; Ikai, A. The importance of being knotted: Effects of the C-terminal knot structure on enzymatic and mechanical properties of bovine carbonic anhydrase II. FEBS Lett. 2002, 519, 35–40. [Google Scholar] [CrossRef]
- Dzubiella, J. Sequence-specific size, structure, and stability of tight protein knots. Biophys. J. 2009, 96, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Makarov, D.E. Translocation of a knotted polypeptide through a pore. J. Chem. Phys. 2008, 129, 121107. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Zhang, H.; Tempczyk, A.; Krajewski, W.; Bonander, N.; Toedt, J.; Howard, A.; Eisenstein, E.; Herzberg, O. Structure of the YibK methyltransferase from Haemophilus influenzae (HI0766): A cofactor bound at a site formed by a knot. Proteins 2003, 51, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Nureki, O.; Shirouzu, M.; Hashimoto, K.; Ishitani, R.; Terada, T.; Tamakoshi, M.; Oshima, T.; Chijimatsu, M.; Takio, K.; Vassylyev, D.G.; et al. An enzyme with a deep trefoil knot for the active-site architecture. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1129–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowska, J.I.; Noel, J.K.; Onuchic, J.N. Energy landscape of knotted protein folding. Proc. Natl. Acad. Sci. USA 2012, 109, 17783–17788. [Google Scholar] [CrossRef] [PubMed]
- Virnau, P.; Mirny, L.A.; Kardar, M. Intricate knots in proteins: Function and evolution. PLoS Comput. Biol. 2006, 2, e122. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Brunzelle, J.S.; Forest, K.T.; Vierstra, R.D. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 2005, 438, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, F.; Lim, N.C.; Mandal, S.S.; Pelz, B.; Ng, W.P.; Schlierf, M.; Jackson, S.E.; Rief, M. Knotting and unknotting of a protein in single molecule experiments. Proc. Natl. Acad. Sci. USA 2016, 113, 7533–7538. [Google Scholar] [CrossRef] [PubMed]
- Mallam, A.L.; Rogers, J.M.; Jackson, S.E. Experimental detection of knotted conformations in denatured proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8189–8194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Yang, L.; Liu, P.; Gao, Y.Q.; Zhao, X.S. Single-molecule detection reveals knot sliding in TrmD denaturation. Chemistry 2013, 19, 5909–5916. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, O.V.; Stepanenko, O.V.; Kuznetsova, I.M.; Verkhusha, V.V.; Turoverov, K.K. Sensitivity of superfolder GFP to ionic agents. PLoS ONE 2014, 9, e110750. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, I.M.; Stepanenko, O.V.; Turoverov, K.K.; Zhu, L.; Zhou, J.M.; Fink, A.L.; Uversky, V.N. Unraveling multistate unfolding of rabbit muscle creatine kinase. Biochim. Biophys. Acta 2002, 1596, 138–155. [Google Scholar] [CrossRef]
- Bushmarina, N.A.; Kuznetsova, I.M.; Biktashev, A.G.; Turoverov, K.K.; Uversky, V.N. Partially folded conformations in the folding pathway of bovine carbonic anhydrase II: A fluorescence spectroscopic analysis. Chembiochem 2001, 2, 813–821. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Stepanenko, O.V.; Stepanenko, O.V.; Povarova, O.I.; Biktashev, A.G.; Verkhusha, V.V.; Shavlovsky, M.M.; Turoverov, K.K. The place of inactivated actin and its kinetic predecessor in actin folding-unfolding. Biochemistry 2002, 41, 13127–13132. [Google Scholar] [CrossRef] [PubMed]
- Staiano, M.; Scognamiglio, V.; Rossi, M.; D’Auria, S.; Stepanenko, O.V.; Kuznetsova, I.M.; Turoverov, K.K. Unfolding and refolding of the glutamine-binding protein from Escherichia coli and its complex with glutamine induced by guanidine hydrochloride. Biochemistry 2005, 44, 5625–5633. [Google Scholar] [CrossRef] [PubMed]
- Staiano, M.; D’Auria, S.; Varriale, A.; Rossi, M.; Marabotti, A.; Fini, C.; Stepanenko, O.V.; Kuznetsova, I.M.; Turoverov, K.K. Stability and dynamics of the porcine odorant-binding protein. Biochemistry 2007, 46, 11120–11127. [Google Scholar] [CrossRef] [PubMed]
- Seward, H.E.; Bagshaw, C.R. The photochemistry of fluorescent proteins: Implications for their biological applications. Chem. Soc. Rev. 2009, 38, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, O.V.; Fonin, A.V.; Stepanenko, O.V.; Morozova, K.S.; Verkhusha, V.V.; Kuznetsova, I.M.; Turoverov, K.K.; Staiano, M.; D’Auria, S. New insight in protein-ligand interactions. 2. Stability and properties of two mutant forms of the D-galactose/D-glucose-binding protein from E. coli. J. Phys. Chem. B 2011, 115, 9022–9032. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Yoshida, A.; Oikawa, H. Hypothesis: Structural heterogeneity of the unfolded proteins originating from the coupling of the local clusters and the long-range distance distribution. Biophys. Rev. 2018, 10, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Phillips, K.J.; Liu, D.R. Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 2007, 129, 10110–10112. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, O.V.; Kuznetsova, I.M.; Turoverov, K.K.; Huang, C.; Wang, C.C. Conformational change of the dimeric DsbC molecule induced by GdnHCl. A study by intrinsic fluorescence. Biochemistry 2004, 43, 5296–5303. [Google Scholar] [CrossRef] [PubMed]
- Povarova, O.I.; Kuznetsova, I.M.; Turoverov, K.K. Differences in the pathways of proteins unfolding induced by urea and guanidine hydrochloride: Molten globule state and aggregates. PLoS ONE 2010, 5, e15035. [Google Scholar] [CrossRef] [PubMed]
- Meuzelaar, H.; Panman, M.R.; Woutersen, S. Guanidinium-Induced Denaturation by Breaking of Salt Bridges. Angew. Chem. 2015, 54, 15255–15259. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.P.; Dima, R.I.; Brooks, B.; Thirumalai, D. Interactions between hydrophobic and ionic solutes in aqueous guanidinium chloride and urea solutions: Lessons for protein denaturation mechanism. J. Am. Chem. Soc. 2007, 129, 7346–7353. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, P.; Bhattacharyya, R. Geometry of nonbonded interactions involving planar groups in proteins. Prog. Biophys. Mol. Biol. 2007, 95, 83–137. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Tanford, C. Acid-base titrations in concentrated guanidine hydrochloride. Dissociation constants of the guamidinium ion and of some amino acids. J. Am. Chem. Soc. 1967, 89, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.J.; Harris, D.R.; Mills, H.H. The crystal structure of guanidinium chloride. Acta Crystallogr. 1965, 19, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Bolen, D.W.; Rose, G.D. Structure and energetics of the hydrogen-bonded backbone in protein folding. Annu. Rev. Biochem. 2008, 77, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Mason, P.E.; Neilson, G.W.; Dempsey, C.E.; Barnes, A.C.; Cruickshank, J.M. The hydration structure of guanidinium and thiocyanate ions: Implications for protein stability in aqueous solution. Proc. Natl. Acad. Sci. USA 2003, 100, 4557–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.; Mukherjee, D.; Chen, J.; Gai, F. Do guanidinium and tetrapropylammonium ions specifically interact with aromatic amino acid side chains? Proc. Natl. Acad. Sci. USA 2017, 114, 1003–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shortle, D. The expanded denatured state: An ensemble of conformations trapped in a locally encoded topological space. Adv. Protein Chem. 2002, 62, 1–23. [Google Scholar] [PubMed]
- Toal, S.; Schweitzer-Stenner, R. Local order in the unfolded state: Conformational biases and nearest neighbor interactions. Biomolecules 2014, 4, 725–773. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Malinovska, L.; Saha, S.; Wang, J.; Alberti, S.; Krishnan, Y.; Hyman, A.A. ATP as a biological hydrotrope. Science 2017, 356, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Hamiaux, C.; Prange, T.; Ries-Kautt, M.; Ducruix, A.; Lafont, S.; Astier, J.P.; Veesler, S. The decameric structure of bovine pancreatic trypsin inhibitor (BPTI) crystallized from thiocyanate at 2.7 A resolution. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Saludjian, P.; Prange, T.; Navaza, J.; Menez, R.; Guilloteau, J.P.; Ries-Kautt, M.; Ducruix, A. Structure determination of a dimeric form of erabutoxin-b, crystallized from a thiocyanate solution. Acta. Crystallogr. B 1992, 48, 520–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shcherbakova, D.M.; Verkhusha, V.V. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nat. Methods 2013, 10, 751–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Kuznetsova, I.M. Intrinsic fluorescence of actin. J. Fluoresc. 2003, 13, 41–57. [Google Scholar] [CrossRef]
- Kuznetsova, I.M.; Sulatskaya, A.I.; Povarova, O.I.; Turoverov, K.K. Reevaluation of ANS Binding to Human and Bovine Serum Albumins: Key Role of Equilibrium Microdialysis in Ligand-Receptor Binding Characterization. PLoS ONE 2012, 7, e40845. [Google Scholar] [CrossRef] [PubMed]
- Fonin, A.V.; Sulatskaya, A.I.; Kuznetsova, I.M.; Turoverov, K.K. Fluorescence of dyes in solutions with high absorbance. Inner filter effect correction. PLoS ONE 2014, 9, e103878. [Google Scholar] [CrossRef] [PubMed]
- Turoverov, K.K.; Biktashev, A.G.; Dorofeiuk, A.V.; Kuznetsova, I.M. A complex of apparatus and programs for the measurement of spectral, polarization and kinetic characteristics of fluorescence in solution. Tsitologiia 1998, 40, 806–817. [Google Scholar] [PubMed]
- Turoverov, K.K.; Verkhusha, V.V.; Shavlovsky, M.M.; Biktashev, A.G.; Povarova, O.I.; Kuznetsova, I.M. Kinetics of actin unfolding induced by guanidine hydrochloride. Biochemistry 2002, 41, 1014–1019. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stepanenko, O.V.; Stepanenko, O.V.; Kuznetsova, I.M.; Turoverov, K.K. The Pathways of the iRFP713 Unfolding Induced by Different Denaturants. Int. J. Mol. Sci. 2018, 19, 2776. https://doi.org/10.3390/ijms19092776
Stepanenko OV, Stepanenko OV, Kuznetsova IM, Turoverov KK. The Pathways of the iRFP713 Unfolding Induced by Different Denaturants. International Journal of Molecular Sciences. 2018; 19(9):2776. https://doi.org/10.3390/ijms19092776
Chicago/Turabian StyleStepanenko, Olesya V., Olga V. Stepanenko, Irina M. Kuznetsova, and Konstantin K. Turoverov. 2018. "The Pathways of the iRFP713 Unfolding Induced by Different Denaturants" International Journal of Molecular Sciences 19, no. 9: 2776. https://doi.org/10.3390/ijms19092776