Quantitative Proteomics of Potato Leaves Infected with Phytophthora infestans Provides Insights into Coordinated and Altered Protein Expression during Early and Late Disease Stages

Abstract
:
1. Introduction
2. Results and Discussion
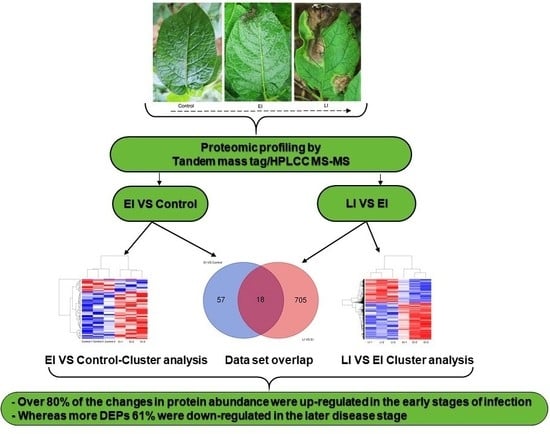
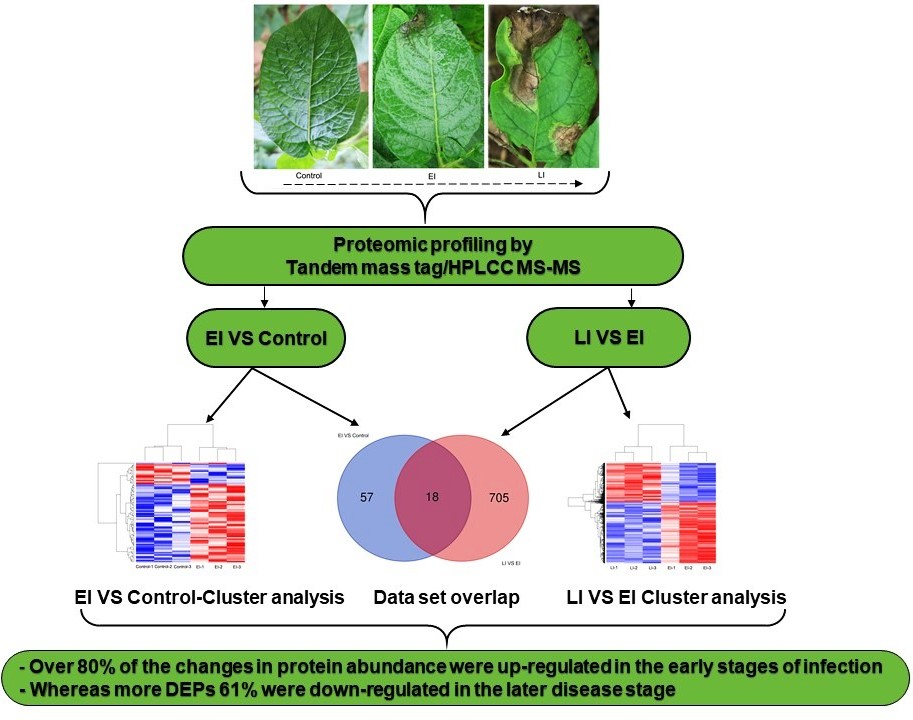
2.1. Subsection Phenotypic Differences between the Three Stages of Disease Conditions
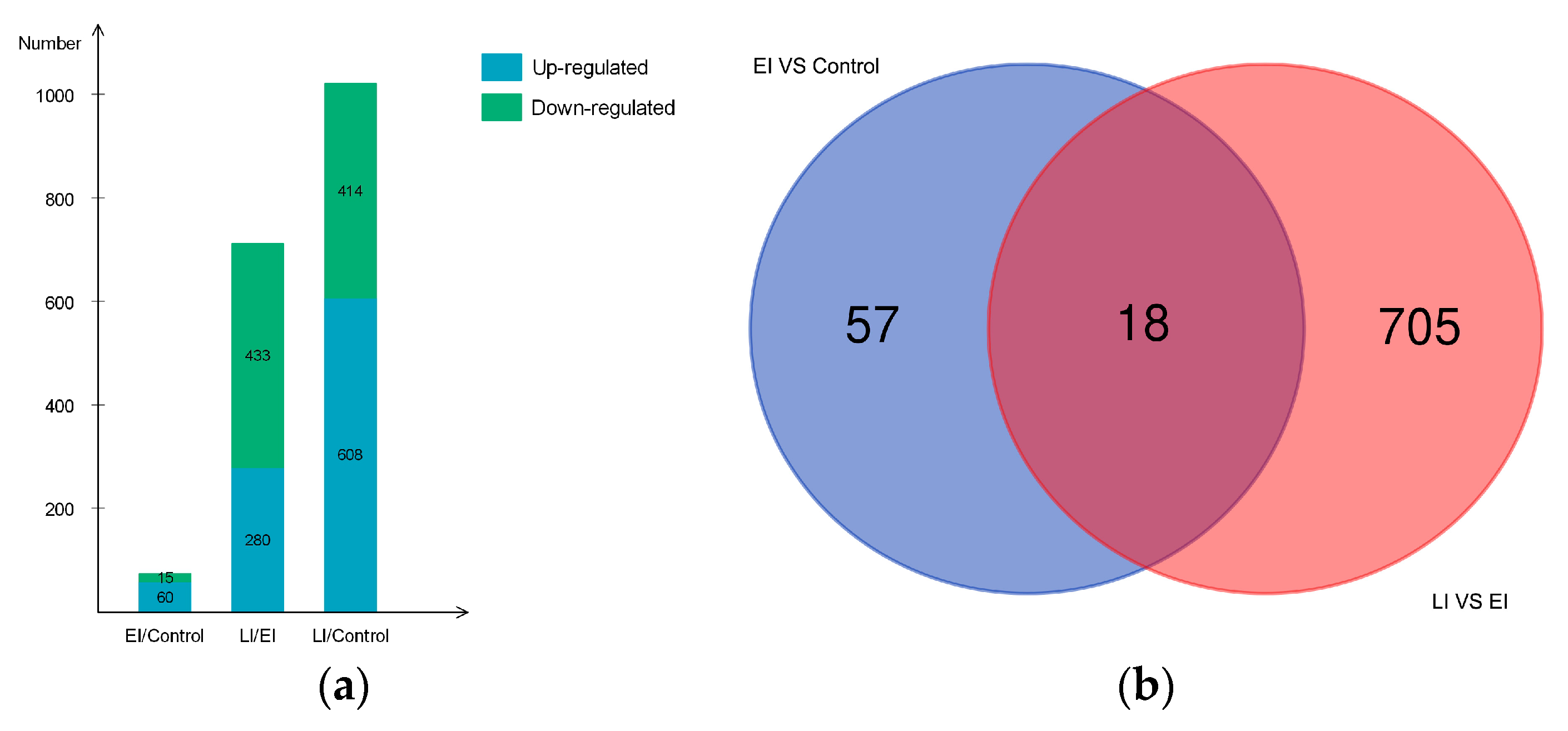
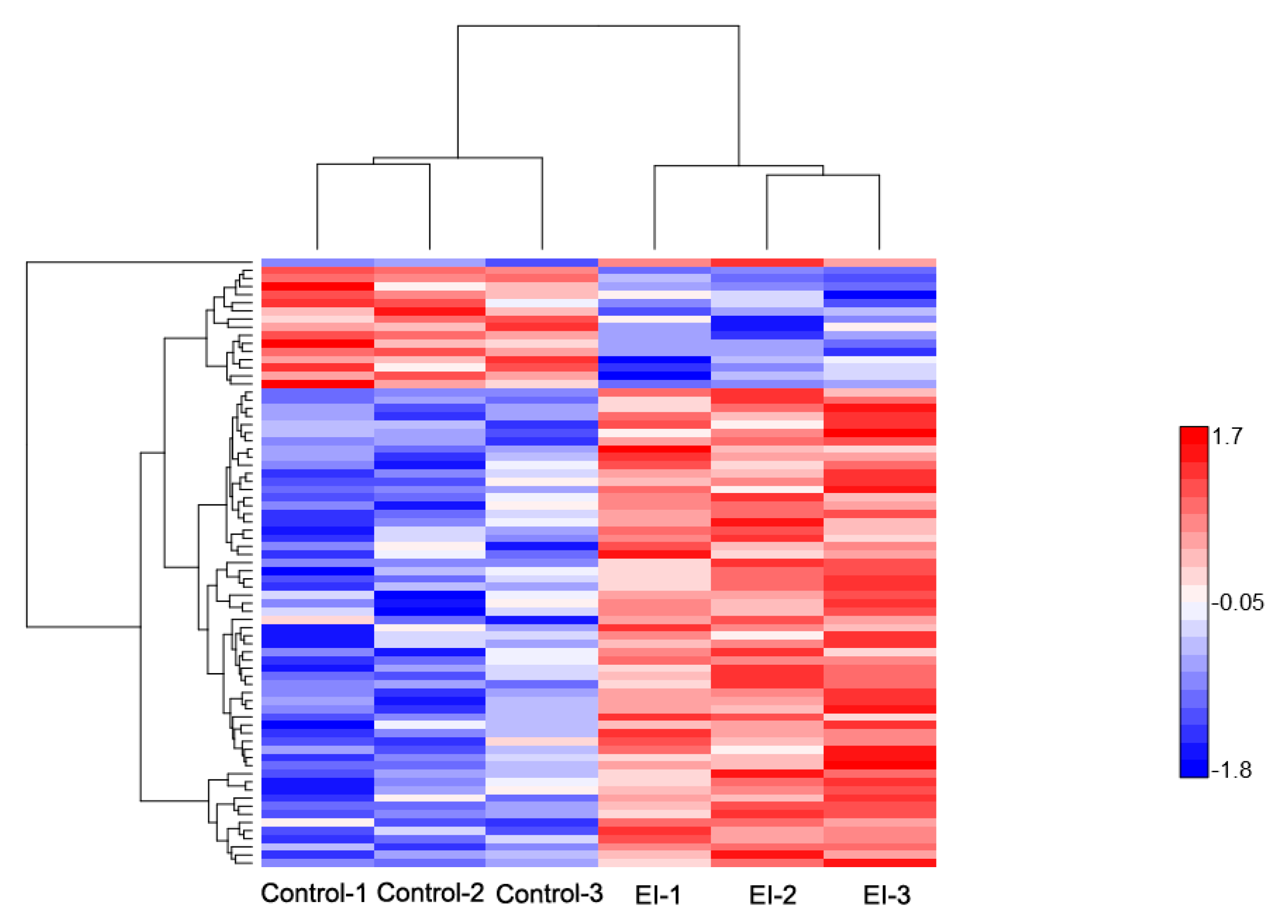
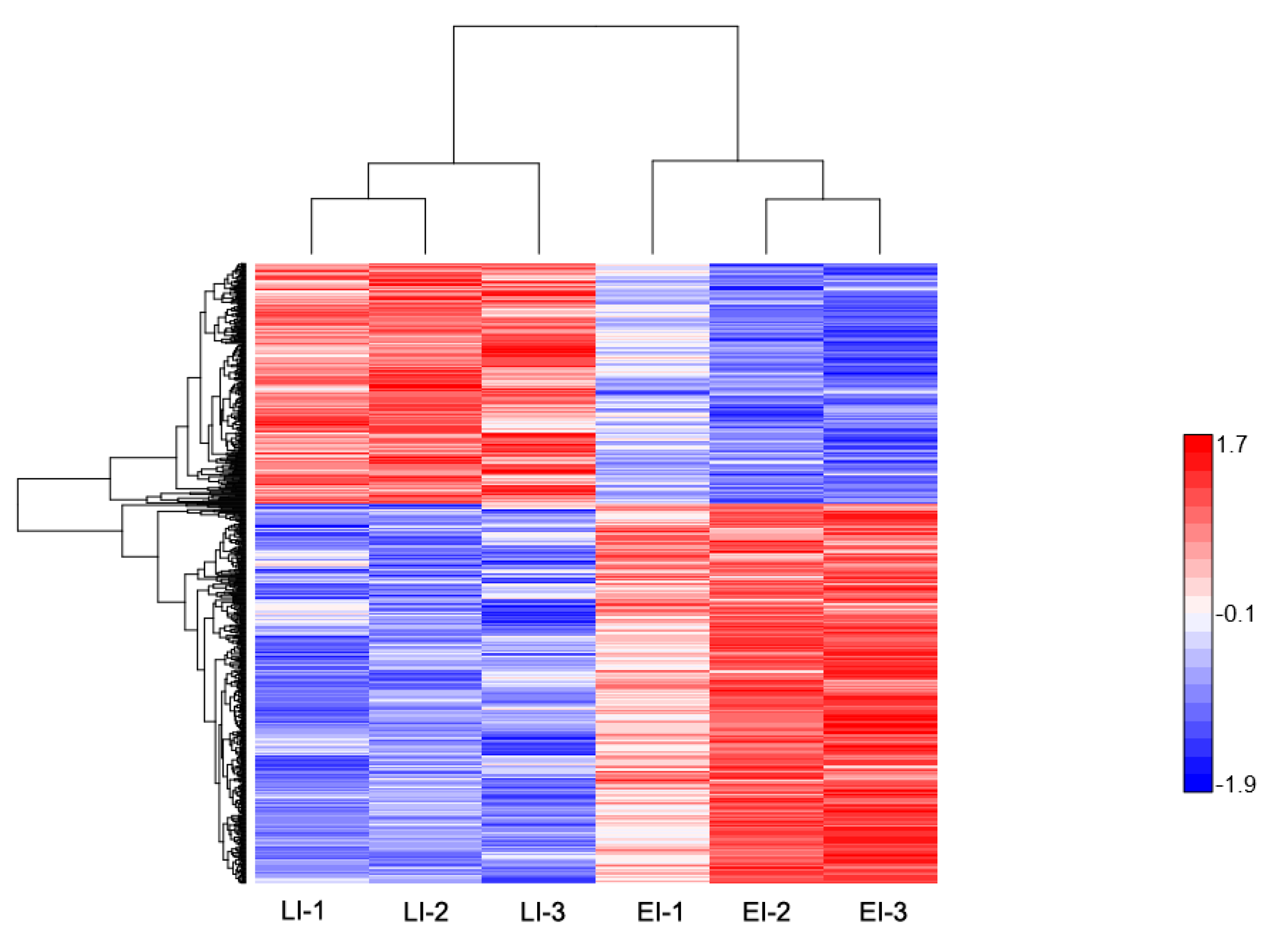
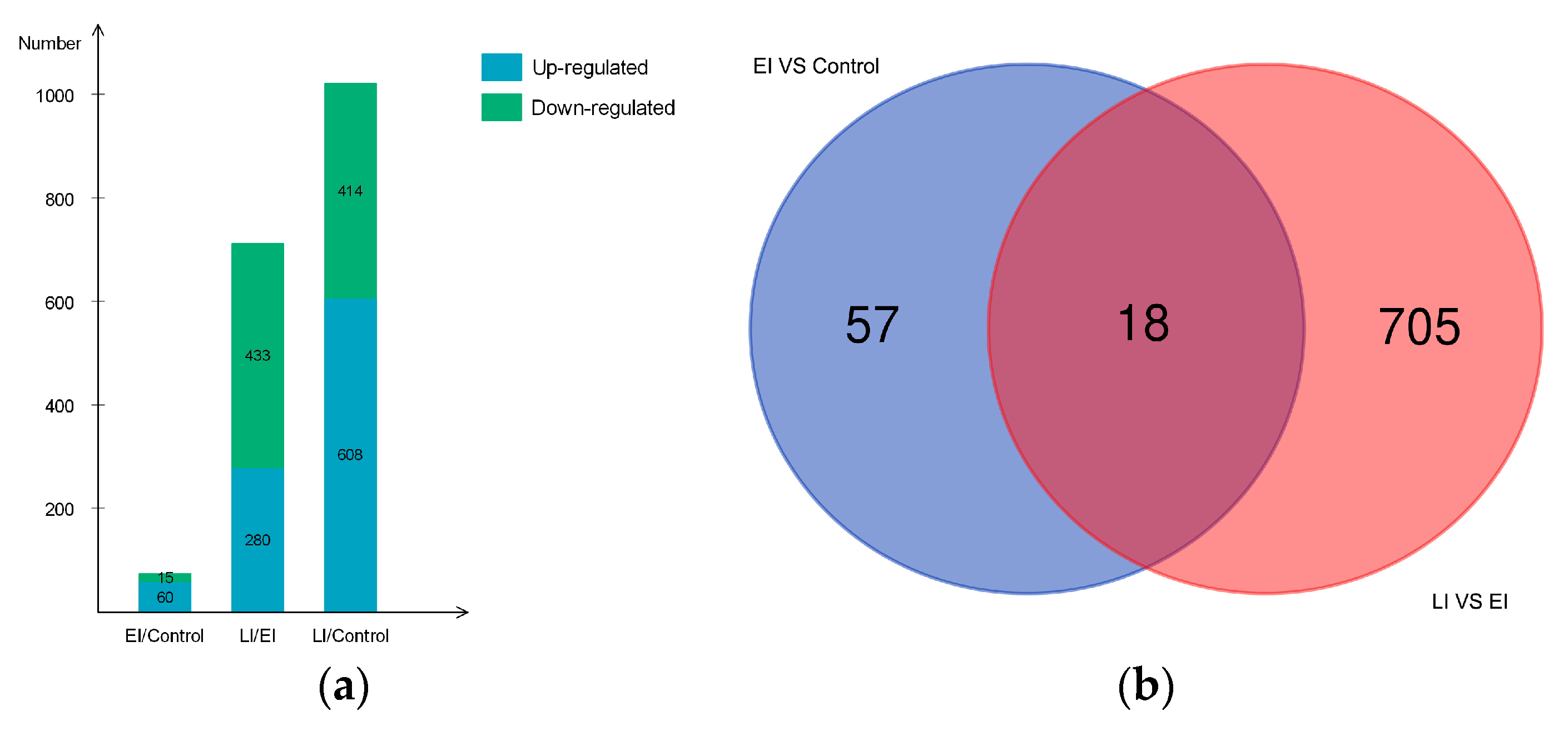
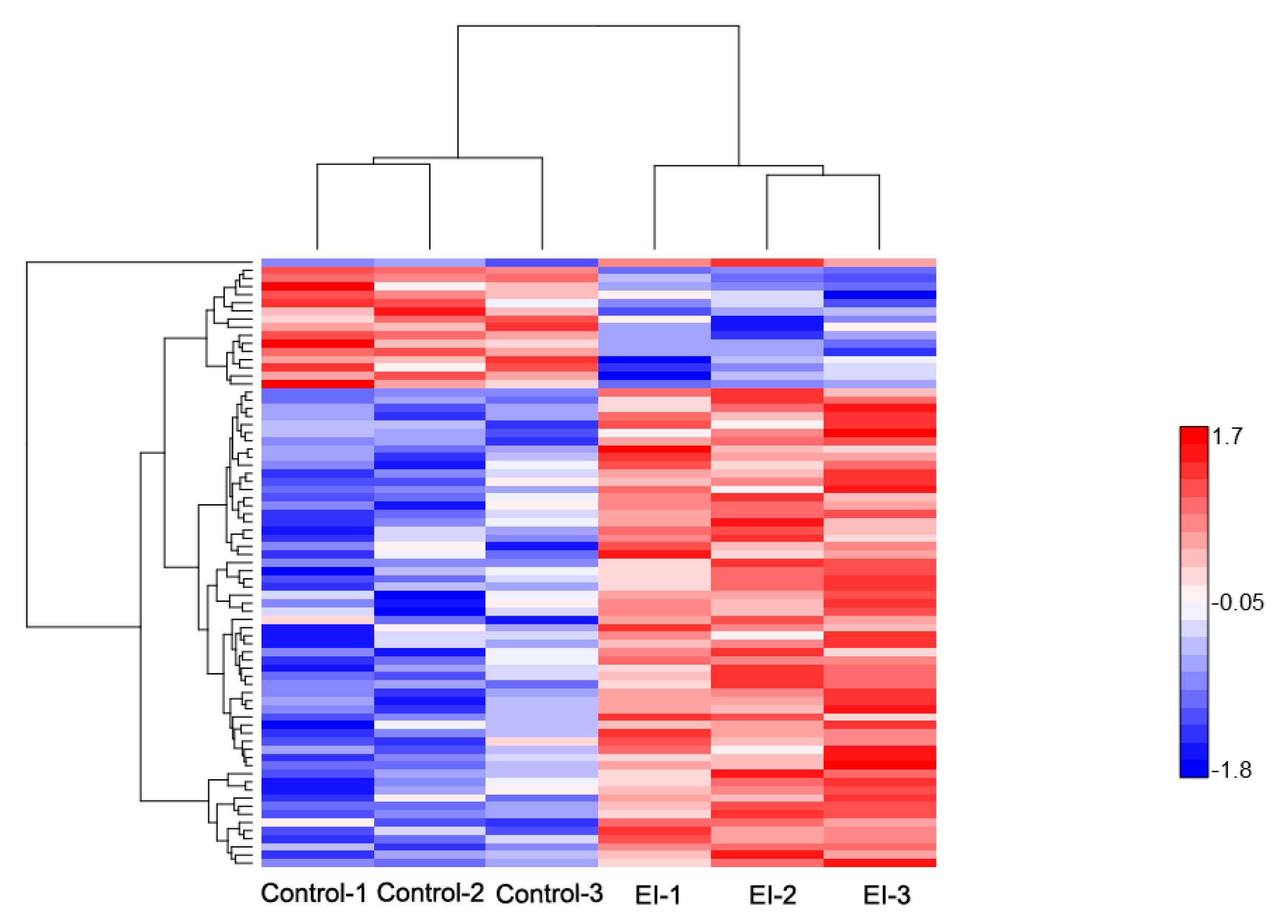
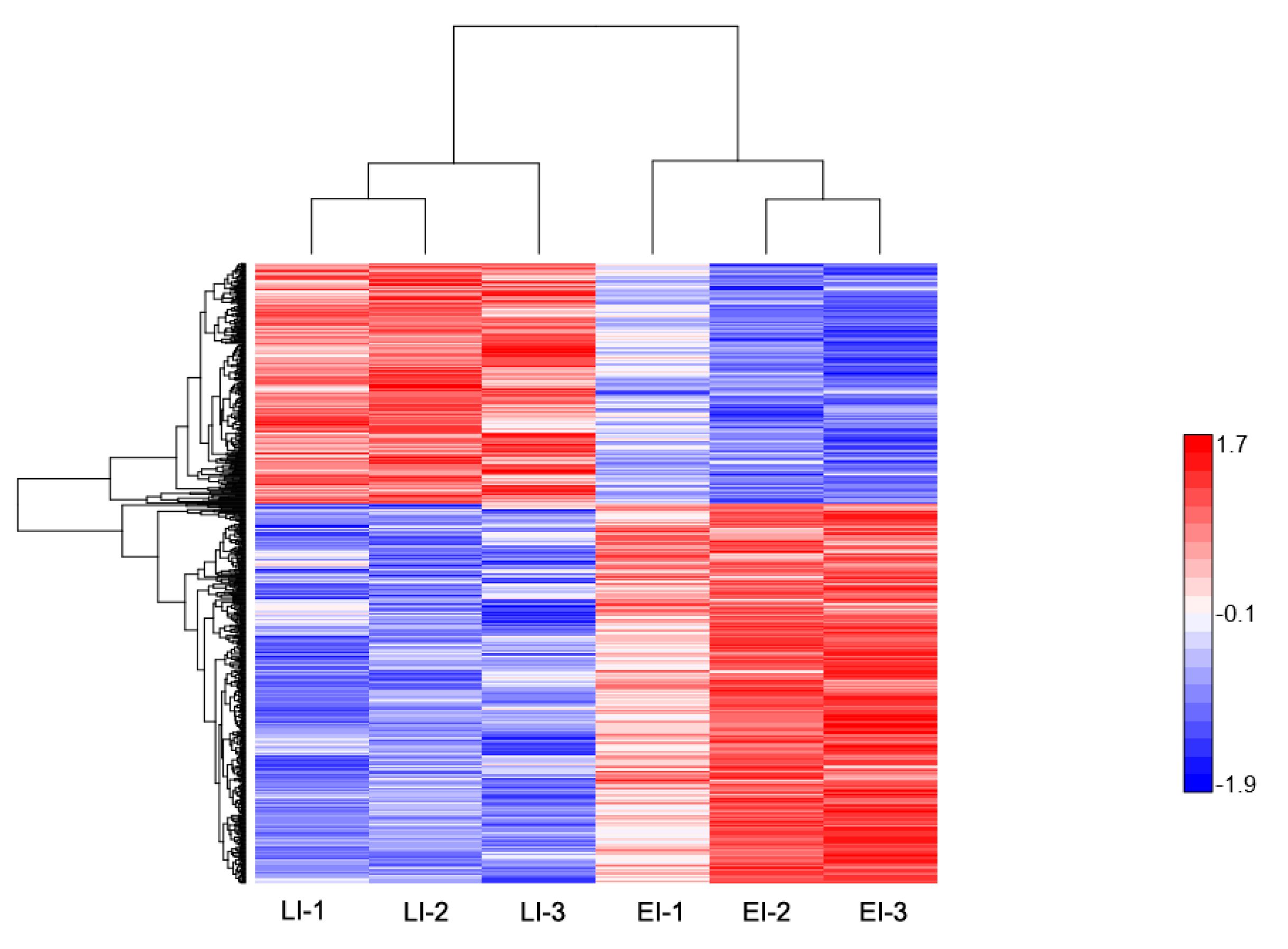
2.2. Overview of Protein Expression in Potato Leaves Challenged with P. infestans Oomycete
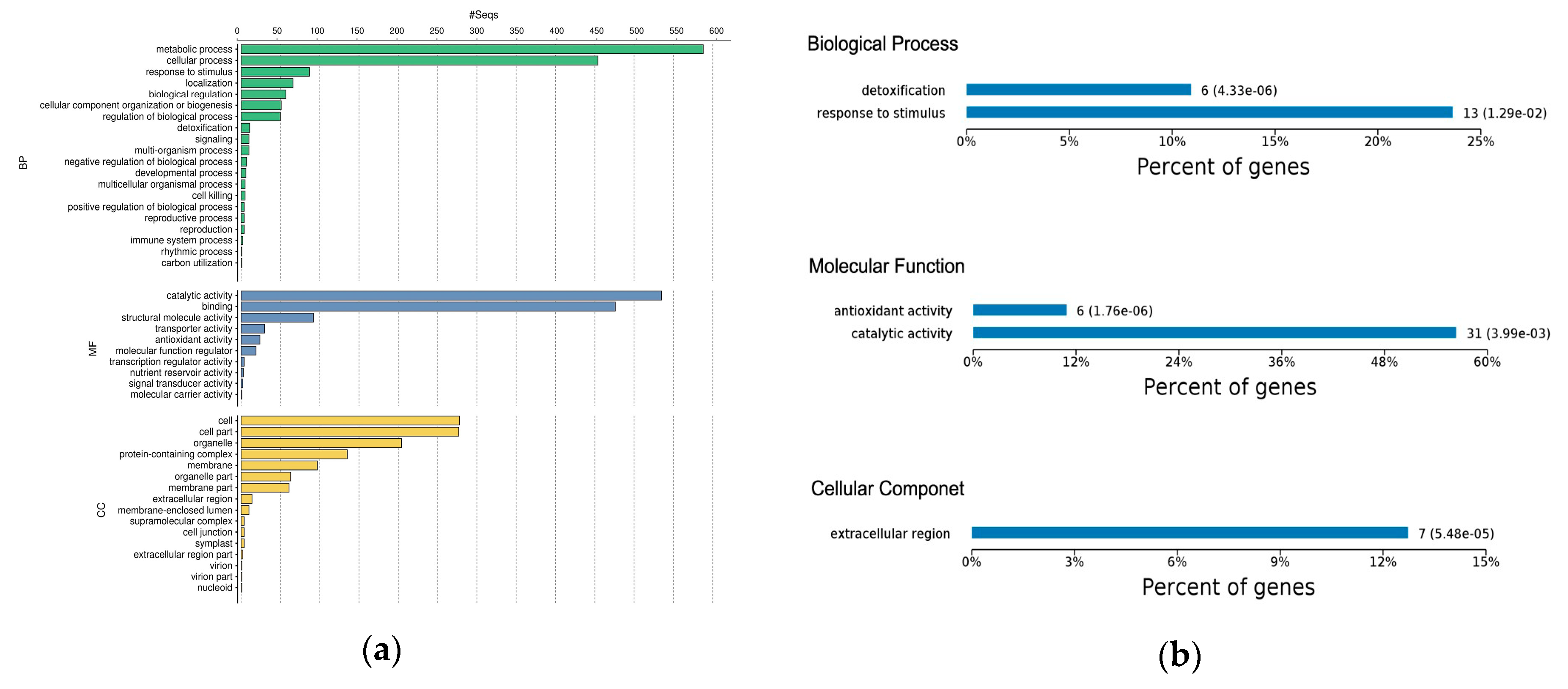
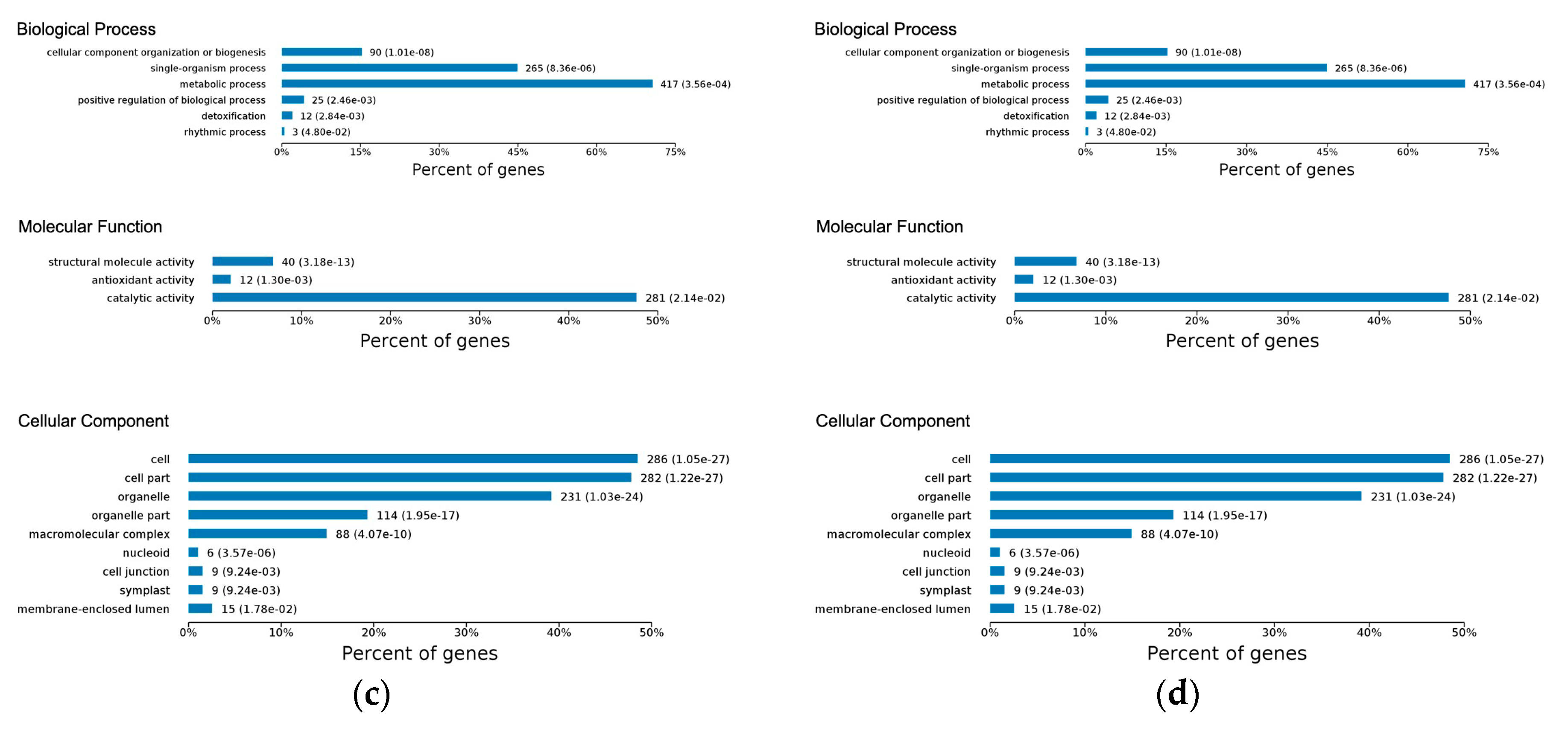
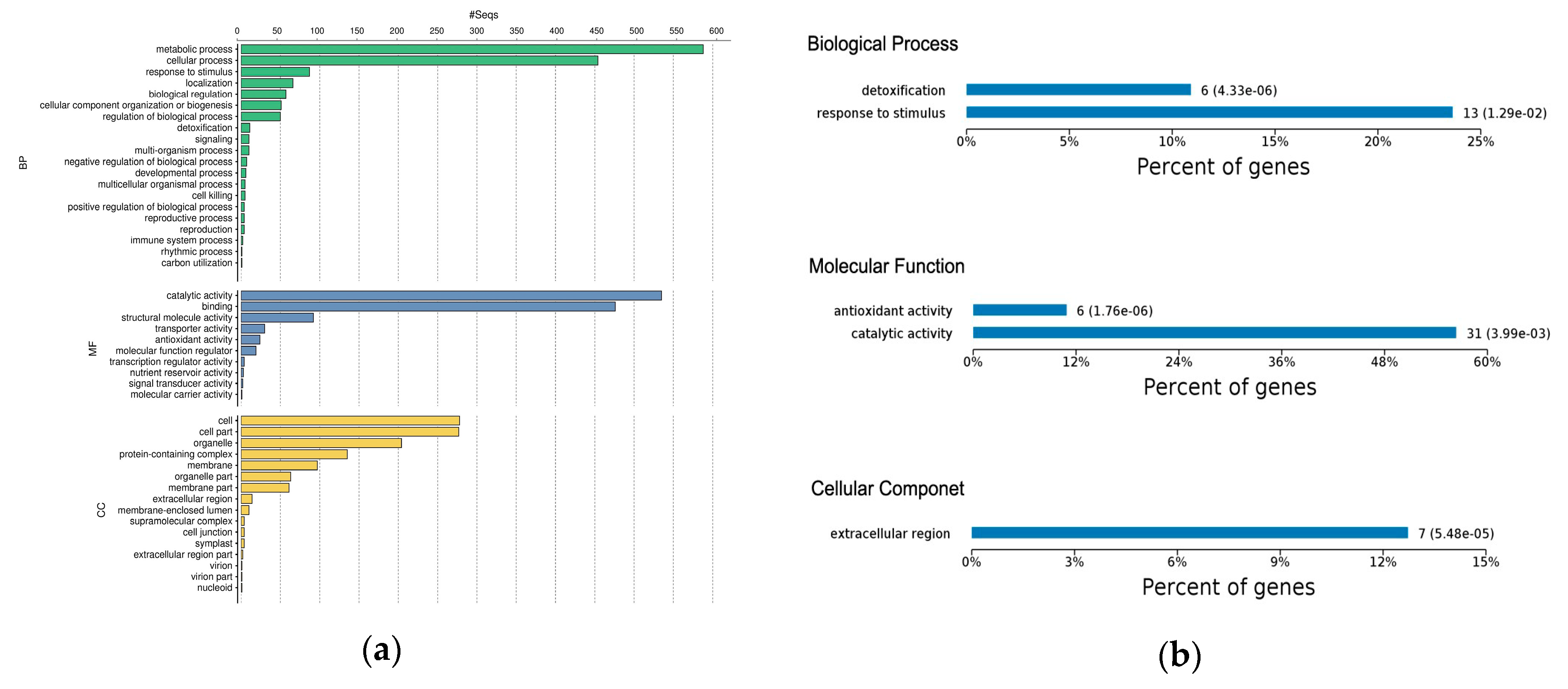
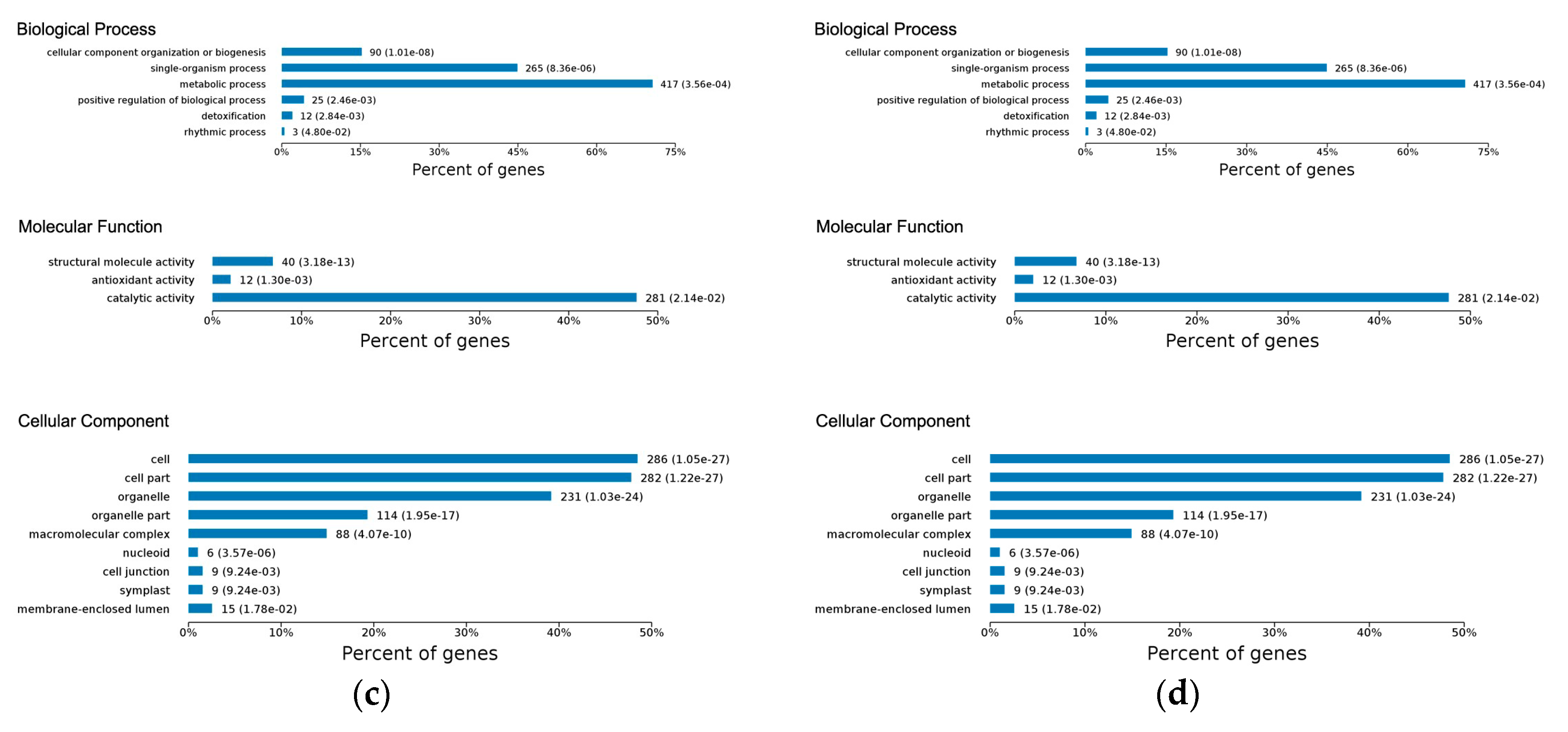
2.3. Gene Ontology Classification of Differentially Expressed Proteins
2.4. Differential Expression Pattern of Proteins Involved at the Early and Late Disease Stages
2.4.1. Early Disease Stage Response Proteins
2.4.2. Late Disease Stage Response Proteins
2.4.3. Differential Regulation of Commonly Shared Proteins in Response to P. infestans during Early and Late Disease Stages
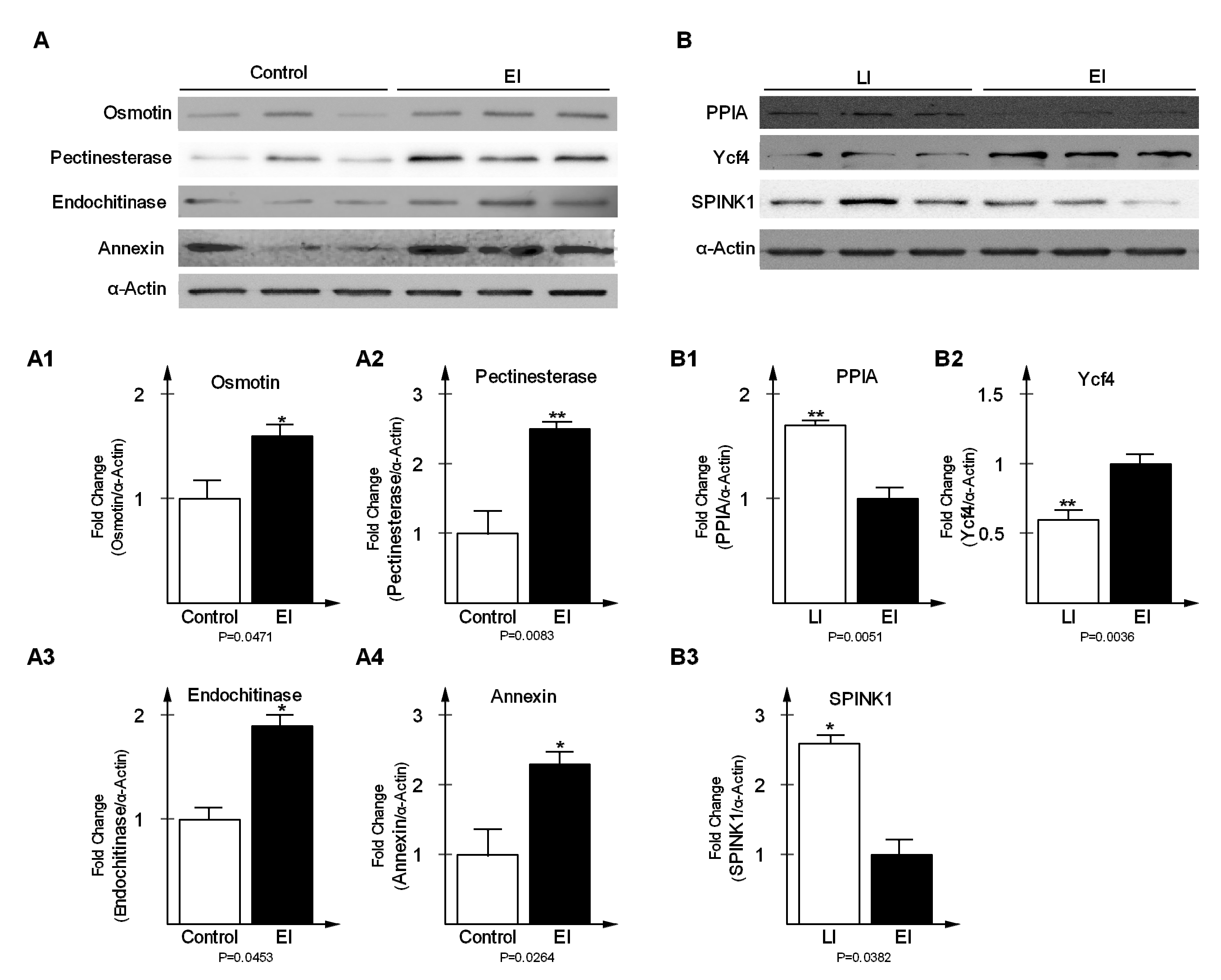
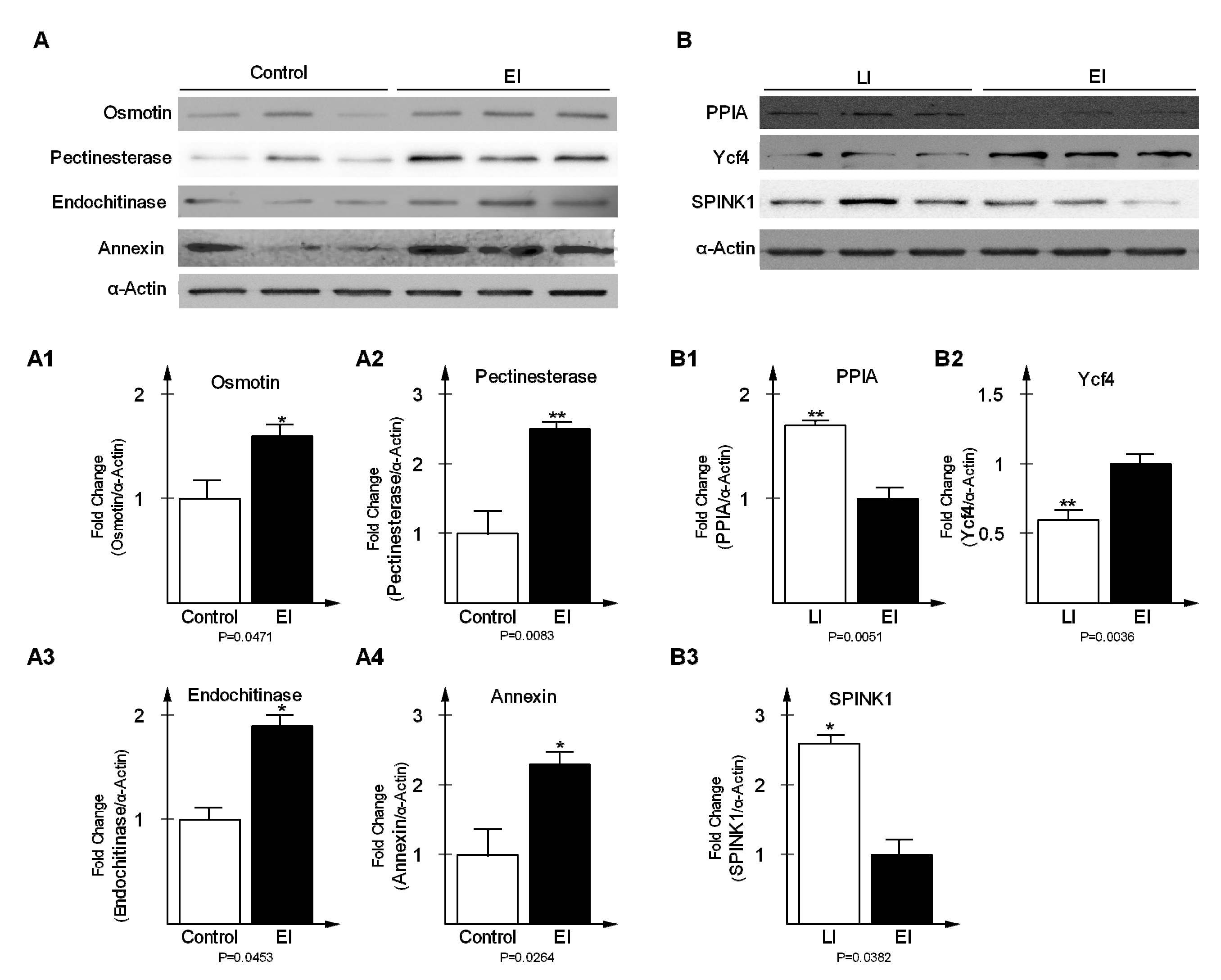
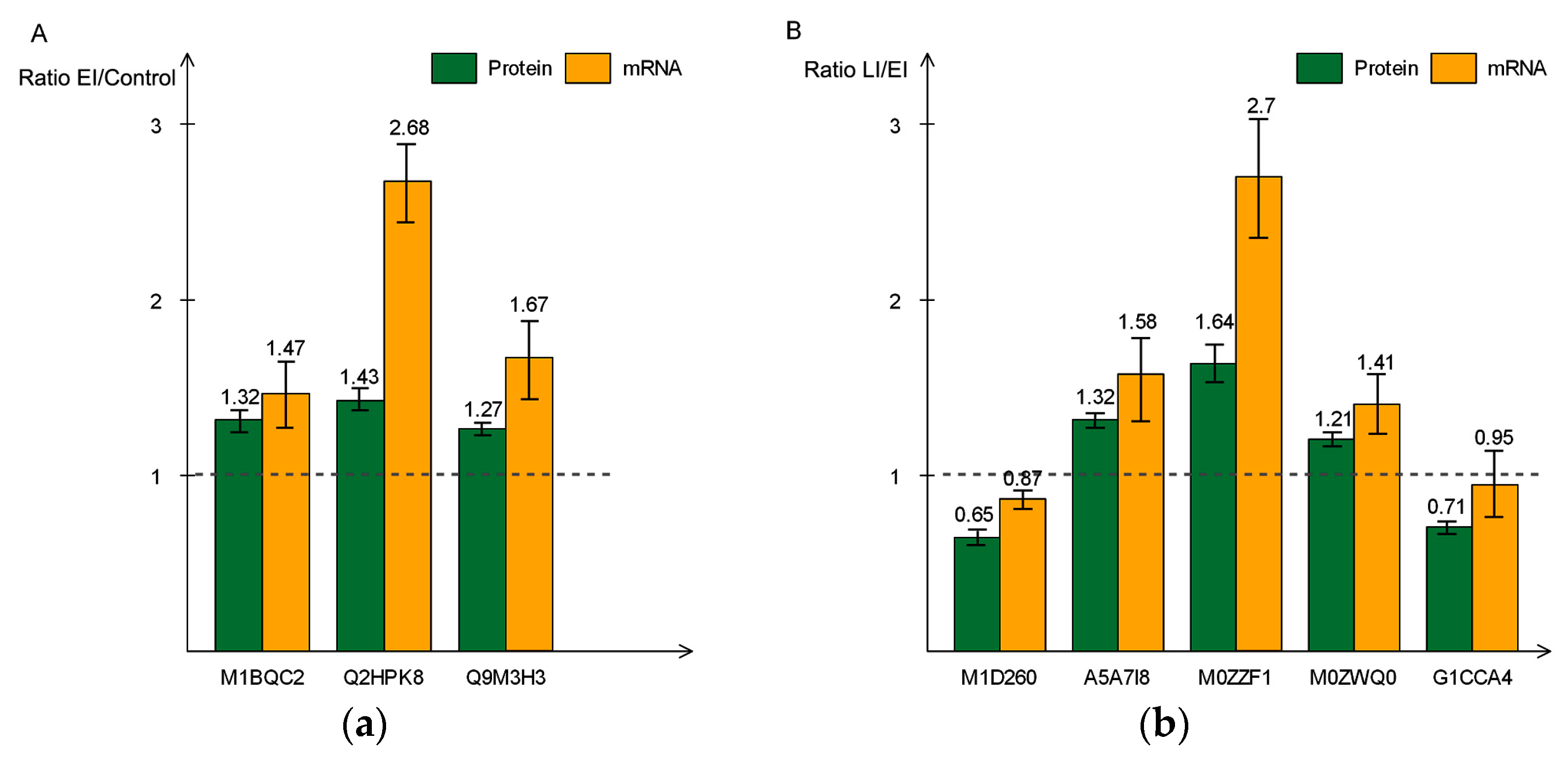
2.5. Validation of Differentially-Expressed Proteins in Early and Late Disease Stages
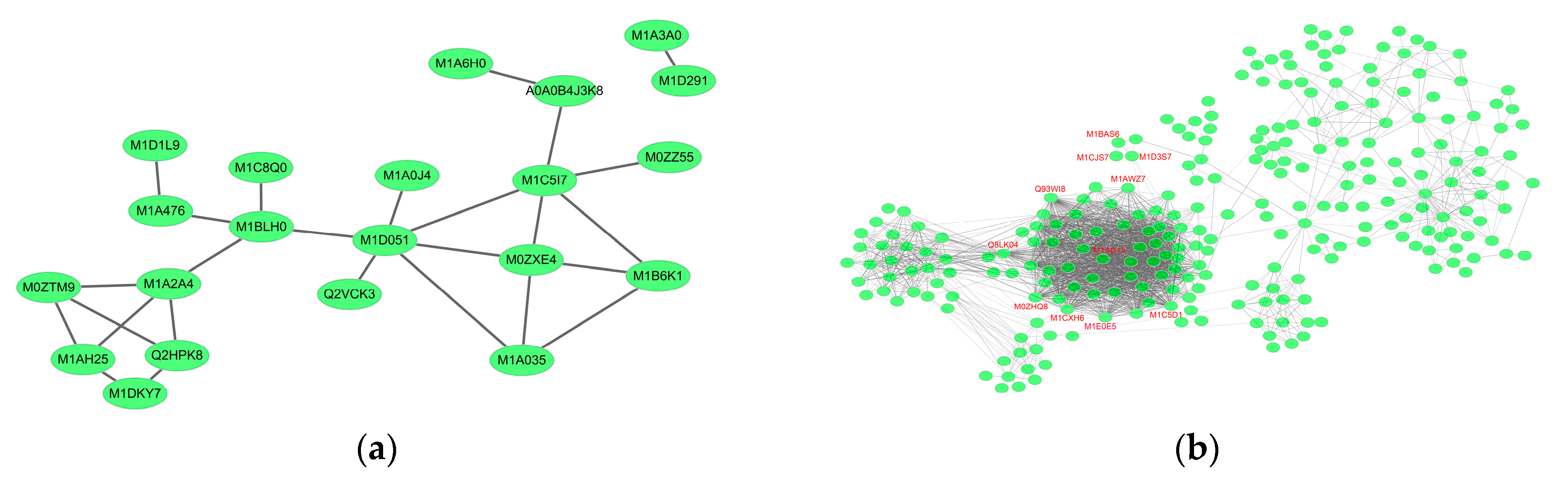
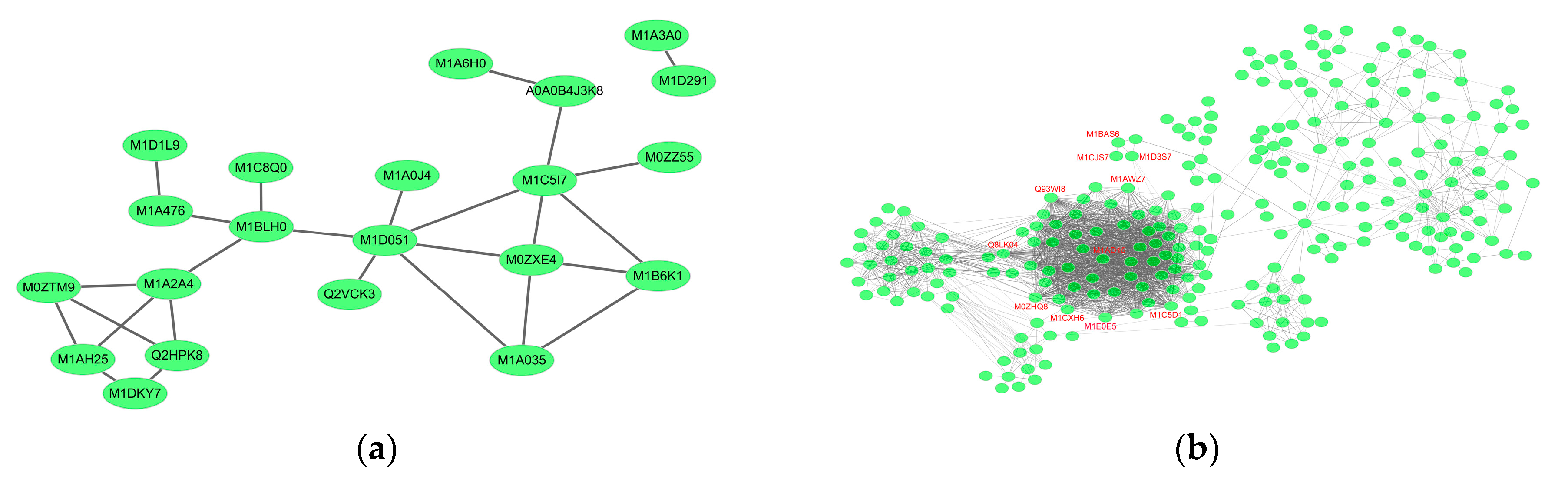
2.6. Essential Proteins for Early and Late Disease Stages
2.6.1. Cv. Sarpo Mira Protein–Protein Interaction during the Early Stages of Infection
2.6.2. Cv. Sarpo Mira Protein–Protein Interaction during the Late Stages of Infection
2.6.3. Correlation Analysis of Protein Expression and mRNA by Real Time-qPCR
3. Materials and Methods
3.1. Plant Material and Growth Conditions
3.2. Protein Extraction
3.3. Protein Digestion
3.4. TMT Labeling and LC-MS/MS Analysis
3.5. Database Search, Protein Identification, and Quantification
3.6. Bioinformatics and Statistical Analysis
3.7. Antibodies and Western Blot Analysis
3.8. Criteria for Selecting Essential Proteins
3.9. Correlation between mRNA and Protein Levels by Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Song, J.; Bradeen, J.M.; Naess, S.K.; Raasch, J.A.; Wielgus, S.M.; Haberlach, G.T.; Lui, J.; Kuang, H.; Austin-Phillips, S.; Buell, C.R.; et al. Gene RB cloned from Solanum bulbocastanum confers broad spectrum resistance to potato late blight. Proc. Natl. Acad. Sci. USA 2003, 100, 9128–9133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Kamoun, S.; Zody, M.C.; Jiang, R.H.Y.; Handsaker, R.E.; Cano, L.M.; et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 2009, 461, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Widmark, A.K.; Andersson, B.; Cassel-Lundhagen, A.; Sandström, M.; Yuen, J.E. Phytophthora infestans in a single field in southwest Sweden early in spring: Symptoms, spatial distribution, and genotypic variation. Plant Pathol. 2007, 56, 573–579. [Google Scholar] [CrossRef]
- Zuluaga, A.P.; Vega-Arreguín, J.C.; Fei, Z.; Ponnala, L.; Lee, S.J.; Matas, A.J.; Patev, S.; Fry, W.E.; Rose, J.K. Transcriptional dynamics of Phytophthora infestans during sequential stages of hemibiotrophic infection of tomato. Mol. Plant Pathol. 2016, 17, 29–41. [Google Scholar] [CrossRef]
- Kaku, H.; Nishizawa, Y.; Ishii-Minami, N.; Akimoto-Tomiyama, C.; Dohmae, N.; Takio, K.; Minami, E.; Shibuya, N. Plant cells recognize chitin fragments for defense signaling through a plasma membrane receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 11086–11091. [Google Scholar] [CrossRef] [Green Version]
- Hayafune, M.; Berisio, R.; Marchetti, R.; Silipo, A.; Kayama, M.; Desaki, Y.; Arima, S.; Squeglia, F.; Ruggiero, A.; Tokuyasu, K.; et al. Chitin-induced activation of immune signaling by the rice receptor CEBiP relies on a unique sandwich-type dimerization. Proc. Natl. Acad. Sci. USA 2014, 111, E404–E413. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Nakano, T.; Takamizawa, D.; Desaki, Y.; Ishii-Minami, N.; Nishizawa, Y.; Minami, E.; Okada, K.; Yamane, H.; Kaku, H.; et al. Two LysM receptor molecules, CEBiP and OsCERK1, cooperatively regulate chitin elicitor signaling in rice. Plant J. 2010, 64, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Miya, A.; Albert, P.; Shinya, T.; Desaki, Y.; Ichimura, K.; Shirasu, K.; Narusaka, Y.; Kawakami, N.; Kaku, H.; Shibuya, N. CERK1, a LysM receptor kinase, is essential for chitin elicitor signaling in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 19613–19618. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Liu, Z.; Song, C.; Hu, Y.; Han, Z.; She, J.; Fan, F.; Wang, J.; Jin, C.; Chang, J.; et al. Chitin-induced dimerization activates a plant immune receptor. Science 2012, 336, 1160–1164. [Google Scholar] [CrossRef]
- Wan, J.; Zhang, X.C.; Neece, D.; Ramonell, K.M.; Clough, S.; Kim, S.Y.; Stacey, M.G.; Stacey, G. A LysM receptor-like kinase plays a critical role in chitin signaling and fungal resistance in Arabidopsis. Plant Cell 2008, 20, 471–481. [Google Scholar] [CrossRef]
- Petutschnig, E.K.; Jones, A.M.E.; Serazetdinova, L.; Lipka, U.; Lipka, V. The lysin motif receptor-like kinase (LysM-RLK) CERK1 is a major chitin-binding protein in Arabidopsis thaliana and subject to chitin-induced phosphorylation. J. Biol. Chem. 2010, 285, 28902–28911. [Google Scholar] [CrossRef] [PubMed]
- Narusaka, Y.; Shinya, T.; Narusaka, M.; Motoyama, N.; Shimada, H.; Murakami, K.; Shibuya, N. Presence of LYM2 dependent but CERK1 independent disease resistance in Arabidopsis. Plant Signal. Behav. 2013, 8, e25345. [Google Scholar] [CrossRef] [PubMed]
- Shinya, T.; Motoyama, N.; Ikeda, A.; Wada, M.; Kamiya, K.; Hayafune, M.; Kaku, H.; Shibuya, N. Functional characterization of CEBiP and CERK1 homologs in Arabidopsis and rice reveals the presence of different chitin receptor systems in plants. Plant Cell Physiol. 2012, 53, 1696–1706. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, C.; Petutschnig, E.; Benitez-Alfonso, Y.; Beck, M.; Robatzek, S.; Lipka, V.; Maule, A.J. LYM2-dependent chitin perception limits molecular flux via plasmodesmata. Proc. Natl. Acad. Sci. USA 2013, 110, 9166–9170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Kars, I.; Essenstam, B.; Liebrand, T.W.H.; Wagemakers, L.; Elberse, J.; Tagkalaki, P.; Tjoitang, D.; van den Ackerveken, G.; van Kan, J.A. Fungal endopolygalacturonases are recognized as microbe-associated molecular patterns by the Arabidopsis receptor-like protein responsiveness to botrytis polygalacturonases1. Plant Physiol. 2014, 164, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Ron, M.; Avni, A. The receptor for the fungal elicitor ethylene-inducing xylanase is a member of a resistance-like gene family in tomato. Plant Cell Am. Soc. Plant Biol. 2004, 16, 1604–1615. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, R.; Van Esse, H.P.; Maruthachalam, K.; Bolton, M.D.; Santhanam, P.; Saber, M.K.; Zhang, Z.; Usami, T.; Lievens, B.; Subbarao, K.V.; et al. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 5110–5115. [Google Scholar] [CrossRef] [Green Version]
- Fradin, E.F.; Zhang, Z.; Ayala, J.C.J.; Castroverde, C.D.M.; Nazar, R.N.; Robb, J.; Liu, C.M.; Thomma, B.P. Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiol. Am. Soc. Plant Biol. 2009, 150, 320–332. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Kamoun, S. A catalog of the effector secretome of plant pathogenic oomycetes. Annu. Rev. Phytopathol. 2006, 44, 41–60. [Google Scholar] [CrossRef]
- Tian, M.; Benedetti, B.; Kamoun, S. A Second Kazal-like protease inhibitor from Phytophthora infestans inhibits and interacts with the apoplastic pathogenesis-related protease P69B of tomato. Plant Physiol. 2005, 138, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurindo, B.S.; Laurindo, R.D.F.; Fontes, P.P.; Vital, C.E.; Delazari, F.T.; Baracat-Pereira, M.C.; da Silva, D.J.H. Comparative analysis of constitutive proteome between resistant and susceptible tomato genotypes regarding to late blight. Funct. Integr. Genom. 2018, 18, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.K.G.; Jorgensen, M.M.; Bennike, T.B.; Stensballe, A. Time-course investigation of Phytophthora infestans infection of potato leaf from three cultivars by quantitative proteomics. Data Br. 2016, 6, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.; Alexandersson, E.; Sandin, M.; Resjö, S.; Lenman, M.; Hedley, P.; Levander, F.; Andreasson, E. Quantitative proteomics and transcriptomics of potato in response to Phytophthora infestans in compatible and incompatible interactions. Bmc Genom. 2014, 15, 497. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.; Dumas-Gaudot, E.; Renaut, J.; Sergeant, K. Gel-Based and Gel-Free Quantitative Proteomics Approaches at a Glance. Int. J. Plant Genom. 2012, 2012, 494572. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Borza, T.; Peters, R.D.; Coffin, R.H.; Al-mughrabi, K.I.; Pinto, D.M.; Wang-Pruski, G. Proteomics analysis suggests broad functional changes in potato leaves triggered by phosphites and a complex indirect mode of action against Phytophthora infestans. J. Proteom. 2013, 93, 207–223. [Google Scholar] [CrossRef]
- Werner, T.; Sweetman, G.; Savitski, M.F.; Mathieson, T.; Bantscheff, M.; Savitski, M.M. Ion coalescence of neutron encoded TMT 10-plex reporter ions. Anal. Chem. 2014, 86, 3594–3601. [Google Scholar] [CrossRef]
- Rietman, H.; Bijsterbosch, G.; Cano, L.M.; Lee, H.R.; Vossen, J.H.; Jacobsen, E.; Visser, R.G.; Kamoun, S.; Vleeshouwers, V.G. Qualitative and quantitative late blight resistance in the potato cultivar Sarpo Mira is determined by the perception of five distinct RXLR effectors. Mol. Plant-Microbe Interact. 2012, 25, 910–919. [Google Scholar] [CrossRef]
- Guo, T.; Wang, X.W.; Shan, K.; Sun, W.; Guo, L.Y. The Loricrin-Like Protein (LLP) of Phytophthora infestans is Required for Oospore Formation and Plant Infection. Front. Plant Sci. 2017, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Mpina, M.H.; Birch, P.R.; Bouwmeester, K.; Govers, F. Phytophthora infestans RXLR Effector AVR1 Interacts with Exocyst Component Sec5 to Manipulate Plant Immunity. Plant Physiol. 2015, 169, 1975–1990. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Yogendra, K.N.; Kumar, A.; Sarkar, K.; Li, Y.; Pushpa, D.; Mosa, K.A.; Duggavathi, R.; Kushalappa, A.C. Transcription factor StWRKY1 regulates phenylpropanoid metabolites conferring late blight resistance in potato. J. Exp. Bot. 2015, 66, 7377–7389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, C.M.; Chapple, C. The phenylpropanoid pathway in Arabidopsis. Arab. Book 2011, 9, E0152. [Google Scholar] [CrossRef]
- Ah-Fong, A.; Shrivastava, J.; Judelson, H.S. Lifestyle, gene gain and loss, and transcriptional remodeling cause divergence in the transcriptomes of Phytophthora infestans and Pythium ultimum during potato tuber colonization. Bmc Genom. 2017, 18, 764. [Google Scholar] [CrossRef]
- Hückelhoven, R. Cell Wall-Associated Mechanisms of Disease Resistance and Susceptibility. Annu. Rev. Phytopathol. 2007, 45, 101–127. [Google Scholar] [CrossRef]
- Sanchez, L.M.; Ohno, Y.; Miura, Y.; Kawakita, K.; Doke, N. Host selective suppression by water-soluble glucans from Phytophthora spp. of hypersensitive cell death of suspension-cultured cells from some solanaceous plants caused by hyphal wall elicitors of the fungi. Ann. Phytopathol. Soc. Jpn. 1992, 58, 664–670. [Google Scholar] [CrossRef]
- Andreu, A.; Tonón, C.; Van Damme, M.; Huarte, M.; Daleo, G. Effect of glucans from different races of Phytophthora infestans on defense reactions in potato tuber. Eur. J. Plant Pathol. 1998, 104, 777–783. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Zhang, H.; Yang, Y.; Ge, X.; Song, F. A rice serine carboxypeptidase-like gene OsBISCPL1 is involved in regulation of defense responses against biotic and oxidative stress. Gene 2008, 420, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Jashni, M.K.; Mehrabi, R.; Collemare, J.; Mesarich, C.H.; de Wit, P.J. The battle in the apoplast: Further insights into the roles of proteases and their inhibitors in plant-pathogen interactions. Front. Plant Sci. 2015, 6, 584. [Google Scholar] [CrossRef] [PubMed]
- Avrova, A.O.; Taleb, N.; Rokka, V.M.; Heilbronn, J.; Campbell, E.; Hein, I.; Gilroy, E.M.; Cardle, L.; Bradshaw, J.E.; Stewart, H.E.; et al. Potato oxysterol binding protein and cathepsin B are rapidly up-regulated in independent defence pathways that distinguish R gene-mediated and field resistances to Phytophthora infestans. Mol. Plant Pathol. 2004, 5, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, S.C.; Kim, M.H.; Lim, H.T.; Park, Y.K.; Hahm, K.S. Antimicrobial activity studies on a trypsin-chymotrypsin protease inhibitor obtained from potato. Biochem. Biophys. Res. Commun. 2005, 330, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.J.; Doerner, P.W.; Zhu, Q.; Lamb, C.J. Isolation of a monocot 3-hydroxy-3-methylglutaryl coenzyme A reductase gene that is elicitor-inducible. Plant Mol. Biol. 1994, 25, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.B.; Pagano, M.R.; Daleo, G.R.; Guevara, M.G. Hydrophobic proteins secreted into the apoplast may contribute to resistance against Phytophthora infestans in potato. Plant Physiol. Biochem. 2012, 60, 59–66. [Google Scholar] [CrossRef]
- Roumeliotis, E.; Kloosterman, B.; Oortwijn, M.; Visser, R.G.; Bachem, C.W. The PIN family of proteins in potato and their putative role in tuberization. Front. Plant Sci. 2013, 4, 524. [Google Scholar] [CrossRef]
- Torto-Alalibo, T.A.; Tripathy, S.; Smith, B.M.; Arredondo, F.D.; Zhou, L.; Li, H.; Chibucos, M.C.; Qutob, D.; Gijzen, M.; Mao, C.; et al. Expressed sequence tags from Phytophthora sojae reveal genes specific to development and infection. Mol. Plant-Microbe Interact. 2007, 20, 781–793. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, Y.; Wu, R.; Jiang, D.; Bai, B.; Tan, D.; Yan, T.; Sun, X.; Zhang, Q.; Wu, Z. Antibacterial Activity of Juglone against Staphylococcus aureus: From Apparent to Proteomic. Int. J. Mol. Sci. 2016, 17, 965. [Google Scholar] [CrossRef]
- Eliot, A.C.; Kirsch, J.F. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu. Rev. Biochem. 2004, 73, 383–415. [Google Scholar] [CrossRef]
- Van Damme, M.; Bozkurt, T.O.; Cakir, C.; Schornack, S.; Sklenar, J.; Jones, A.M.; Kamoun, S. The Irish Potato Famine Pathogen Phytophthora infestans Translocates the CRN8 Kinase into Host Plant Cells. PloS Pathog. 2012, 8, E1002875. [Google Scholar] [CrossRef] [PubMed]
- Bretz, J.R.; Mock, N.M.; Charity, J.C.; Zeyad, S.; Baker, C.J.; Hutcheson, S.W. A translocated protein tyrosine phosphatase of Pseudomonas syringae pv. tomato DC3000 modulates plant defence response to infection. Mol. Microbiol. 2003, 49, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underwood, W.; Zhang, S.; He, S.Y. The Pseudomonas syringae type III effector tyrosine phosphatase HopAO1 suppresses innate immunity in Arabidopsis thaliana. Plant J. 2007, 52, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Rivas, S.; Rougon-Cardoso, A.; Smoker, M.; Schauser, L.; Yoshioka, H.; Jones, J.D.G. CITRX thioredoxin interacts with the tomato Cf-9 resistance protein and negatively regulates defense. EMBO J. 2004, 23, 2156–2165. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, V.; Ludwig, A.A.; Jones, J.D.G. CITRX thioredoxin is a putative adaptor protein connecting Cf-9 and the ACIK1 protein kinase during the Cf-9/Avr9-induced defense response. Febs Lett. 2006, 580, 4236–4241. [Google Scholar] [CrossRef] [PubMed]
- Burra, D.D.; Vetukuri, R.R.; Resjö, S.; Grenville-Briggs, L.J.; Andreasson, E. RNAseq and Proteomics for Analysing Complex Oomycete Plant Interactions. Curr. Issues Mol. Biol. 2016, 19, 73–88. [Google Scholar] [PubMed]
- Akino, S.; Takemoto, D.; Hosaka, K. Phytophthora infestans: A review of past and current studies on potato late blight. J. Gen. Plant Pathol. 2014, 80, 24–37. [Google Scholar] [CrossRef]
- Shibata, Y.; Kawakita, K.; Takemoto, D. Age-related resistance of Nicotiana benthamiana against hemibiotrophic pathogen Phytophthora infestans requires both ethylene- and salicylic acid-mediated signaling pathways. Mol. Plant Microbe Interact. 2010, 23, 1130–1142. [Google Scholar] [CrossRef]
- D’Ovidio, R.; Mattei, B.; Roberti, S.; Bellincampi, D. Polygalacturonases, polygalacturonase-inhibiting proteins and pectic oligomers in plant-pathogen interactions. Biochim. Biophys. Acta 2004, 1696, 237–244. [Google Scholar] [CrossRef]
- Diener, A.C.; Ausubel, F.M. Resistance to Fusarium Oxysporum 1, a dominant Arabidopsis disease-resistance gene, is not race specific. Genetics 2005, 171, 305–321. [Google Scholar] [CrossRef]
- Brutus, A.; Sicilia, F.; Macone, A.; Cervone, F.; De Lorenzo, G. A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc. Natl. Acad. Sci. USA 2010, 107, 9452–9457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boller, T.; Felix, G. A Renaissance of Elicitors: Perception of microbe-associated molecular patterns and danger signals by pattern-recognition receptors. Annu. Rev. Plant Biol. 2009, 60, 379–407. [Google Scholar] [CrossRef] [PubMed]
- Le Berre, J.Y.; Engler, G.; Panabières, F. Exploration of the late stages of the tomato–Phytophthora parasitica interactions through histological analysis and generation of expressed sequence tags. New Phytol. 2008, 177, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, D.; Furuse, K.; Doke, N.; Kawakita, K. Identification of Chitinase and Osmotin-Like Protein as Actin-Binding Proteins in Suspension-Cultured Potato Cells. Plant Cell Physiol. 1997, 38, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Vleeshouwers, V.G.A.A.; Dooijeweert, W.V.; Govers, F.; Kamoun, S.; TColona, L.T. Does basal PR gene expression in Solanum species contribute to non-specific resistance to Phytophthora infestans? Physiol. Mol. Plant Pathol. 2000, 57, 35–42. [Google Scholar] [CrossRef]
- Draffehn, A.M.; Li, L.; Krezdorn, N.; Ding, J.; Lübeck, J.; Strahwald, J.; Muktar, M.S.; Walkemeier, B.; Rotter, B.; Gebhardt, C. Comparative transcript profiling by SuperSAGE identifies novel candidate genes for controlling potato quantitative resistance to late blight not compromised by late maturity. Front. Plant Sci. 2013, 4, 423. [Google Scholar] [CrossRef]
- Mosquera, T.; Alvarez, M.F.; Jiménez-Gómez, J.M.; Muktar, M.S.; Paulo, M.J.; Steinemann, S.; Li, J.; Draffehn, A.; Hofmann, A.; Lübeck, J.; et al. Targeted and Untargeted Approaches Unravel Novel Candidate Genes and Diagnostic SNPs for Quantitative Resistance of the Potato (Solanum tuberosum L.) to Phytophthora infestans Causing the Late Blight Disease. PLoS ONE 2016, 11, E0156254. [Google Scholar] [CrossRef]
- Bass, S.; Gu, Q.; Christen, A. Multicopy suppressors of prc mutant Escherichia coli include two HtrA (DegP) protease homologs (HhoAB), DksA, and a truncated R1pA. J. Bacteriol. 1996, 178, 1154–1161. [Google Scholar] [CrossRef]
- Whisson, S.C.; Boevink, P.C.; Wang, S.; Birch, P.R.J. The cell biology of late blight disease. Curr. Opin. Microbiol. 2016, 34, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Zhang, Q.; Zhang, M.; Gu, B.; Huang, G.; Wang, Q.; Shan, W. The protein disulfide isomerase 1 of Phytophthora parasitica (PpPDI1) is associated with the haustoria-like structures and contributes to plant infection. Front. Plant Sci. 2015, 6, 632. [Google Scholar] [CrossRef]
- Oliveira, A.E.; Gomes, V.M.; Sales, M.P.; Fernandes, K.V.; Carlini, C.R.; Xavier-Filho, J. The toxicity of jack bean [Canavalia ensiformis (L.) DC.] canatoxin to plant pathogenic fungi. Rev. Bras. Biol. 1999, 59, 59–62. [Google Scholar] [CrossRef]
- Becker-Ritta, A.B.; Martinelli, H.S.; Mitidieri, S.; Feder, V.; Wassermann, G.E.; Santi, L.; Vainstein, M.H.; Oliveira, J.T.A.; Fiuza, L.M.; Pasquali, G.; et al. Antifungal activity of plant and bacterial ureases. Toxicon 2007, 50, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Resjö, S.; Brus, M.; Ali, A.; Meijer, H.J.G.; Sandin, M.; Govers, F.; Levander, F.; Grenville-Briggs, L.; Andreasson, E. Proteomic Analysis of Phytophthora infestans Reveals the Importance of Cell Wall Proteins in Pathogenicity. Mol. Cell Proteom. 2017, 16, 1958–1971. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, A.; Kondo, Y.; Tanaka, H.; Matsumura, I.; Ishibashi, T.; Sudo, T.; Satoh, Y.; Yokota, T.; Ezoe, S.; Oritani, K.; et al. An Anti-Apoptotic Molecule, Anamorsin, Functions in Both Iron-Sulfur Protein Assembly and Cellular Iron Homeostasis. Blood 2011, 118, 2106. [Google Scholar]
- Gyetvai, G.; Sonderkaer, M.; Gobel, U.; Basekow, R.; Ballvora, A.; Imhoff, M.; Kersten, B.; Kare-Lehman Nielsen, K.L.; Gebhardt, C. The Transcriptome of Compatible and Incompatible Interactions of Potato (Solanum tuberosum) with Phytophthora infestans Revealed by DeepSAGE Analysis. PLoS ONE 2012, 7, E31526. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, Y.; Xu, M.; Cheng, Z.; Zhang, D.; Zheng, J. iTRAQ-Based Quantitative Proteomics Analysis of Black Rice Grain Development Reveals Metabolic Pathways Associated with Anthocyanin Biosynthesis. PLoS ONE 2016, 7, e0159238. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hüttenhain, R.; Collins, B.; Aebersold, R. Mass spectrometric protein maps for biomarker discovery and clinical research. Expert Rev. Mol. Diagn. 2013, 13, 811–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcaino, J.A.; Csordas, A.; Del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2015, 44, D447–D456. [Google Scholar] [CrossRef] [Green Version]
- Consortium TGO. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases, and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2010, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.Y.; Wang, H.; Pang, Y.; Xu, C.; Jiao, Y.; Qin, Y.M.; Western, T.L.; Yu, S.X.; Zhu, Y.X. Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fiber and Arabidopsis root hair elongation. Mol. Cell Proteom. 2010, 9, 2019–2033. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Howden, A.J.M.; Stam, R.; Heredia, V.M.; Motion, G.B.; Have, S.T.; Hodge, K.; Amaro, T.M.M.M.; Huitema, E. Quantitative Analysis of the Tomato Nuclear Proteome during Phytophthora Capsici Infection Unveils Regulators of Immunity. New Phytol. 2017, 215, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, G.M.; Podrebarac, D.M.; Greenough, W.T.; Weiler, I.J. The Use of Total Protein Stains as Loading Controls: An Alternative to High-Abundance Single-Protein Controls in Semi-Quantitative Immunoblotting. J. Neurosci. Methods 2008, 172, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Description | Fold Change | p Value | GO Names |
|---|---|---|---|---|
| Early stage up-regulated proteins | ||||
| M0ZZ55 | Uncharacterized protein containing longin N-terminal domains with C-terminal coiled-coil/SNARE motif. | 1.77 | 0.02 | C: integral component of membrane; P: vesicle-mediated transport |
| M0ZTM7 | Osmotin | 1.58 | 0.03 | |
| Q5XUH0 | Osmotin-like protein | 1.54 | 0.002 | |
| M0ZMK7 | Uncharacterized protein with domain named as Bet v 1. Bet v 1 belongs to family 10 of plant pathogenesis-related proteins (PR-10) | 1.51 | 0.004 | P: defense response; P: response to biotic stimulus |
| P52402 | Glucan endo-1,3-beta-glucosidase, basic isoform 3 (Fragment) | 1.45 | 0.03 | F: hydrolase activity, hydrolyzing O-glycosyl compounds; P: carbohydrate metabolic process |
| Q2HPK8 | Putative endochitinase (Fragment) | 1.43 | 0.04 | F: chitinase activity; P: chitin catabolic process; P: cell wall macromolecule catabolic process |
| M1D1L9 | Uncharacterized protein containing domain 2 of Ribosomal protein L2, | 1.36 | 0.02 | F: structural constituent of ribosome; C: ribosome; P: translation |
| M1CUM0 | Uncharacterized protein containing a domain found in the isoprenoid synthase family | 1.34 | 0.04 | F: magnesium ion binding; P: metabolic process; F: terpene synthase activity |
| M1D578 | Peroxidase | 1.34 | 0.04 | F: peroxidase activity; P: response to oxidative stress; F: heme binding; P: hydrogen peroxide catabolic process; P: oxidation-reduction process |
| M1BQC2 | Pectinesterase | 1.32 | 0.03 | F: enzyme inhibitor activity; C: cell wall; F: pectinesterase activity; P: cell wall modification |
| M1A3A0 | Uncharacterized protein containing Pentatricopeptide repeat (PPR) | 1.29 | 0.02 | F: protein binding |
| M1A035 | Carboxypeptidase | 1.28 | 0.03 | F: serine-type carboxypeptidase activity; P: proteolysis |
| M1D051 | Wall-associated kinase | 1.28 | 0.03 | F: protein kinase activity; F: ATP binding; P: protein phosphorylation |
| Q9M3H3 | Annexin | 1.27 | 0.01 | F: calcium ion binding; F: calcium-dependent phospholipid binding |
| M0ZXE4 | Uncharacterized protein contains a domain found in serine peptidases | 1.27 | 0.02 | F: serine-type endopeptidase activity; P: proteolysis |
| Q4FE26 | Proteinase inhibitor 1 PPI3B2 | 1.26 | 0.03 | F: serine-type endopeptidase inhibitor activity; P: response to wounding |
| M1ABL9 | Uncharacterized protein contains AAA+ ATPase domain | 1.25 | 0.003 | F: ATP binding and hydrolysis |
| Q84XQ4 | NtPRp27-like protein | 1.25 | 0.02 | |
| M1A4R1 | Uncharacterized protein with AT hooks a DNA-binding motif | 1.24 | 0.004 | C: nucleosome; F: DNA binding; C: nucleus; P: nucleosome assembly |
| M1C8Q0 | Uncharacterized protein contains Proteasome component Initiation (PCI) domain | 1.22 | 0.02 | F: enzyme regulator activity |
| Early stage down-regulated proteins | ||||
| K7WNX1 | Light-inducible tissue-specific ST-LS1 | 0.83 | 0.001 | C: photosystem II oxygen-evolving complex; P: photosynthesis |
| M0ZJ30 | Uncharacterized protein contains Major facilitator domain | 0.74 | 0.002 | C: membrane; F: transmembrane transporter activity; P: transmembrane transport |
| M1BNJ1 | Uncharacterized protein contains Carotenoid oxygenase binding site. | 0.68 | 0.05 | F: oxidoreductase activity, acting on single donors with incorporation of molecular oxygen, incorporation of two atoms of oxygen; P: oxidation-reduction process |
| Late stage up-regulated proteins | ||||
| M1BD67 | Uncharacterized protein with Acid phosphatase, class B-like domain | 5.68 | 0.03 | F: acid phosphatase activity |
| M0ZTQ4 | Uncharacterized protein predicted with Osmotin/thaumatin-like domain | 5.62 | 0.05 | |
| M1CJS7 | Uncharacterized protein contains CC-NB-ARC and LRR Domains. | 4.00 | 0.01 | F: ADP binding |
| M1B864 | Uncharacterized protein contains Kunitz inhibitor STI-like domain. | 3.65 | 0.04 | F: endopeptidase inhibitor activity |
| M1AU65 | Peroxidase | 2.94 | 0.01 | F: peroxidase activity; P: response to oxidative stress; F: heme binding; P: hydrogen peroxide catabolic process; P: oxidation-reduction process |
| E0WCF2 | PIN-A | 2.92 | 0.0001 | F: serine-type endopeptidase inhibitor activity; P: response to wounding |
| A0A097H185 | PIN-I-Protein | 2.52 | 0.01 | F: serine-type endopeptidase inhibitor activity; P: response to wounding |
| M0ZZF1 | Peptidyl-prolyl cis-trans isomerase | 1.64 | 0.02 | P: protein peptidyl-prolyl isomerization; F: peptidyl-prolyl cis-trans isomerase activity |
| M1BAS6 | Uncharacterized protein (CC-NB-ARC and LRR Domains) | 1.62 | 0.03 | F: ADP binding |
| Q8H9B9 | Hyoscyamine 6-beta-hydroxylase-like protein (Fragment) | 1.62 | 0.03 | F: oxidoreductase activity; P: oxidation-reduction process |
| A5A7I8 | Calcium-dependent protein kinase 5 | 1.32 | 0.04 | F: protein kinase activity; F: calcium ion binding; F: ATP binding; P: protein phosphorylation |
| M1AKD9 | Uncharacterized protein contains a Histidine kinase/HSP90-like ATPase domain | 1.30 | 0.02 | F: ATP binding; P: protein folding; P: response to stress; F: unfolded protein binding |
| Q8VX50 | Putative receptor-like serine-threonine protein kinase | 1.22 | 0.05 | F: protein kinase activity; F: ATP binding; P: protein phosphorylation |
| M1D3S7 | Uncharacterized protein contains a CC-NB-ARC and LRR Domain. | 1.22 | 0.05 | F: ADP binding |
| M0ZWQ0 | V-type proton ATPase proteolipid subunit | 1.21 | 0.03 | F: proton transmembrane transporter activity; P: ATP hydrolysis coupled proton transport; C: proton-transporting V-type ATPase, V0 domain |
| Late stage down-regulated proteins | ||||
| K7WNV9 | Thioredoxin | 0.82 | 0.05 | P: glycerol ether metabolic process; F: protein disulfide oxidoreductase activity; P: cell redox homeostasis |
| G1CCA4 | Photosystem I assembly protein Ycf4 | 0.71 | 0.03 | C: photosystem I; P: photosynthesis; C: integral component of membrane |
| M1D260 | FK506-binding protein | 0.63 | 0.03 | P: histone peptidyl-prolyl isomerization; F: peptidyl-prolyl cis-trans isomerase activity; F:FK506 binding; C: nucleolus |
| M1B8N6 | NADPH-protochlorophyllide oxidoreductase | 0.37 | 0.04 | F: protochlorophyllide reductase activity; P: oxidation-reduction process |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, C.; Gao, J.; Zhang, Y.; Wang, Z.; Zhang, D.; Chen, Q.; Ye, X.; Xu, Y.; Yang, G.; Yan, L.; et al. Quantitative Proteomics of Potato Leaves Infected with Phytophthora infestans Provides Insights into Coordinated and Altered Protein Expression during Early and Late Disease Stages. Int. J. Mol. Sci. 2019, 20, 136. https://doi.org/10.3390/ijms20010136
Xiao C, Gao J, Zhang Y, Wang Z, Zhang D, Chen Q, Ye X, Xu Y, Yang G, Yan L, et al. Quantitative Proteomics of Potato Leaves Infected with Phytophthora infestans Provides Insights into Coordinated and Altered Protein Expression during Early and Late Disease Stages. International Journal of Molecular Sciences. 2019; 20(1):136. https://doi.org/10.3390/ijms20010136
Chicago/Turabian StyleXiao, Chunfang, Jianhua Gao, Yuanxue Zhang, Zhen Wang, Denghong Zhang, Qiaoling Chen, Xingzhi Ye, Yi Xu, Guocai Yang, Lei Yan, and et al. 2019. "Quantitative Proteomics of Potato Leaves Infected with Phytophthora infestans Provides Insights into Coordinated and Altered Protein Expression during Early and Late Disease Stages" International Journal of Molecular Sciences 20, no. 1: 136. https://doi.org/10.3390/ijms20010136
APA StyleXiao, C., Gao, J., Zhang, Y., Wang, Z., Zhang, D., Chen, Q., Ye, X., Xu, Y., Yang, G., Yan, L., Cheng, Q., Chen, J., & Shen, Y. (2019). Quantitative Proteomics of Potato Leaves Infected with Phytophthora infestans Provides Insights into Coordinated and Altered Protein Expression during Early and Late Disease Stages. International Journal of Molecular Sciences, 20(1), 136. https://doi.org/10.3390/ijms20010136