Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Overview of Heme Oxygenase Metabolites
2. Anti-Inflammatory Role of HO Metabolites in Neuroinflammatory Diseases
2.1. Anti-Inflammatory Role of HO Metabolites in Various Cells
2.1.1. ECs
2.1.2. Pericytes
2.1.3. Astrocytes
2.1.4. Neurons
2.2. Anti-Inflammatory Role of HO Metabolites in Neuroinflammatory Diseases
2.2.1. Stroke
2.2.2. TBI
2.2.3. Diabetic Retinopathy
2.2.4. AD
3. Regenerative Effects of HO Metabolites
3.1. VEGF
3.2. BDNF
3.3. Stromal Cell-Derived Factor-1 (SDF-1)
3.4. NOS/NO
3.5. OPN
4. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ryter, S.W.; Ma, K.C.; Choi, A.M.K. Carbon monoxide in lung cell physiology and disease. Am. J. Physiol. Cell Physiol. 2018, 314, C211–C227. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Pae, H.O.; Park, J.E.; Lee, Y.C.; Woo, J.M.; Kim, N.H.; Choi, Y.K.; Lee, B.S.; Kim, S.R.; Chung, H.T. Heme oxygenase in the regulation of vascular biology: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 14, 137–167. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Zeller, M.; Cottin, Y.; Vergely, C. Redox Functions of Heme Oxygenase-1 and Biliverdin Reductase in Diabetes. Trends Endocrinol. Metab. 2018, 29, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; D’Agata, V.; Calabrese, V.; Pascale, A.; Colombrita, C.; Alkon, D.; Cavallaro, S. Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res. 2002, 954, 51–59. [Google Scholar] [CrossRef]
- Choi, Y.K.; Maki, T.; Mandeville, E.T.; Koh, S.H.; Hayakawa, K.; Arai, K.; Kim, Y.M.; Whalen, M.J.; Xing, C.; Wang, X.; et al. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat. Med. 2016, 22, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Zucker, S.D.; Goessling, W.; Hoppin, A.G. Unconjugated bilirubin exhibits spontaneous diffusion through model lipid bilayers and native hepatocyte membranes. J. Biol. Chem. 1999, 274, 10852–10862. [Google Scholar] [CrossRef] [PubMed]
- Komuro, A.; Tobe, T.; Nakano, Y.; Yamaguchi, T.; Tomita, M. Cloning and characterization of the cDNA encoding human biliverdin-IX alpha reductase. Biochim. Biophys. Acta 1996, 1309, 89–99. [Google Scholar] [CrossRef]
- Tudor, C.; Lerner-Marmarosh, N.; Engelborghs, Y.; Gibbs, P.E.; Maines, M.D. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 2008, 413, 405–416. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2018, in press. [Google Scholar] [CrossRef]
- Mancuso, C.; Perluigi, M.; Cini, C.; De Marco, C.; Giuffrida Stella, A.M.; Calabrese, V. Heme oxygenase and cyclooxygenase in the central nervous system: A functional interplay. J. Neurosci. Res. 2006, 84, 1385–1391. [Google Scholar] [CrossRef]
- Mancuso, C.; Ragazzoni, E.; Tringali, G.; Liberale, I.; Preziosi, P.; Grossman, A.; Navarra, P. Inhibition of heme oxygenase in the central nervous system potentiates endotoxin-induced vasopressin release in the rat. J. Neuroimmunol. 1999, 99, 189–194. [Google Scholar] [CrossRef]
- Mancuso, C.; Preziosi, P.; Grossman, A.B.; Navarra, P. The role of carbon monoxide in the regulation of neuroendocrine function. Neuroimmunomodulation 1997, 4, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Carraway, M.S.; Suliman, H.B. Carbon monoxide, oxidative stress, and mitochondrial permeability pore transition. Free Radic. Biol. Med. 2006, 40, 1332–1339. [Google Scholar] [CrossRef]
- Kapitulnik, J. Bilirubin: An endogenous product of heme degradation with both cytotoxic and cytoprotective properties. Mol. Pharmacol. 2004, 66, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Heme oxygenase-1: Role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Schipper, H.M. Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox. Res. 1999, 1, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Kim, K.W. Blood-neural barrier: Its diversity and coordinated cell-to-cell communication. BMB Rep. 2008, 41, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Jimi, E.; Zeiss, C.; Hayden, M.S.; Ghosh, S. Constitutively active NF-kappaB triggers systemic TNFalpha-dependent inflammation and localized TNFalpha-independent inflammatory disease. Genes Dev. 2010, 24, 1709–1717. [Google Scholar] [CrossRef]
- Choi, S.; Kim, J.; Kim, J.H.; Lee, D.K.; Park, W.; Park, M.; Kim, S.; Hwang, J.Y.; Won, M.H.; Choi, Y.K.; et al. Carbon monoxide prevents TNF-alpha-induced eNOS downregulation by inhibiting NF-kappaB-responsive miR-155-5p biogenesis. Exp. Mol. Med. 2017, 49, e403. [Google Scholar] [CrossRef]
- Motterlini, R.; Otterbein, L.E. The therapeutic potential of carbon monoxide. Nat. Rev. Drug Discov. 2010, 9, 728–743. [Google Scholar] [CrossRef]
- Sonveaux, P.; Jordan, B.F.; Gallez, B.; Feron, O. Nitric oxide delivery to cancer: Why and how? Eur. J. Cancer 2009, 45, 1352–1369. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Furukawa, M.; Matsuoka, Y.; Tooyama, I.; Kimura, H.; Nomura, Y.; Taniguchi, T. In vitro and in vivo induction of heme oxygenase-1 in rat glial cells: Possible involvement of nitric oxide production from inducible nitric oxide synthase. Glia 1998, 22, 138–148. [Google Scholar] [CrossRef]
- Jansen, T.; Daiber, A. Direct Antioxidant Properties of Bilirubin and Biliverdin. Is there a Role for Biliverdin Reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Jansen, T.; Hortmann, M.; Oelze, M.; Opitz, B.; Steven, S.; Schell, R.; Knorr, M.; Karbach, S.; Schuhmacher, S.; Wenzel, P.; et al. Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J. Mol. Cell Cardiol. 2010, 49, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, P.E.; Miralem, T.; Maines, M.D. Characterization of the human biliverdin reductase gene structure and regulatory elements: Promoter activity is enhanced by hypoxia and suppressed by TNF-alpha-activated NF-kappaB. FASEB J. 2010, 24, 3239–3254. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Central nervous system pericytes in health and disease. Nat. Neurosci. 2011, 14, 1398–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Maki, T.; Choi, Y.K.; Miyamoto, N.; Shindo, A.; Liang, A.C.; Ahn, B.J.; Mandeville, E.T.; Kaji, S.; Itoh, K.; Seo, J.H.; et al. A-Kinase Anchor Protein 12 Is Required for Oligodendrocyte Differentiation in Adult White Matter. Stem Cells 2018, 36, 751–760. [Google Scholar] [CrossRef]
- Cao, L.; Blute, T.A.; Eldred, W.D. Localization of heme oxygenase-2 and modulation of cGMP levels by carbon monoxide and/or nitric oxide in the retina. Vis. Neurosci. 2000, 17, 319–329. [Google Scholar] [CrossRef]
- Lee, B.S.; Heo, J.; Kim, Y.M.; Shim, S.M.; Pae, H.O.; Chung, H.T. Carbon monoxide mediates heme oxygenase 1 induction via Nrf2 activation in hepatoma cells. Biochem. Biophys. Res. Commun. 2006, 343, 965–972. [Google Scholar] [CrossRef]
- Kapupara, K.; Wen, Y.T.; Tsai, R.K.; Huang, S.P. Soluble P-selectin promotes retinal ganglion cell survival through activation of Nrf2 signaling after ischemia injury. Cell Death Dis. 2017, 8, e3172. [Google Scholar] [CrossRef] [PubMed]
- Romeo, G.; Liu, W.H.; Asnaghi, V.; Kern, T.S.; Lorenzi, M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes 2002, 51, 2241–2248. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, P.; Yagi, K.; Ohshiro, Y.; He, Z.; Maeno, Y.; Yamamoto-Hiraoka, J.; Rask-Madsen, C.; Chung, S.W.; Perrella, M.A.; King, G.L. Selective regulation of heme oxygenase-1 expression and function by insulin through IRS1/phosphoinositide 3-kinase/Akt-2 pathway. J. Biol. Chem. 2008, 283, 34327–34336. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nikolakopoulou, A.M.; Zhao, Z.; Sagare, A.P.; Si, G.; Lazic, D.; Barnes, S.R.; Daianu, M.; Ramanathan, A.; Go, A.; et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med. 2018, 24, 326–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Chung, W.S. Phagocytic Roles of Glial Cells in Healthy and Diseased Brains. Biomol. Ther. (Seoul) 2018, 26, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K. A positive circuit of VEGF increases Glut-1 expression by increasing HIF-1alpha gene expression in human retinal endothelial cells. Arch. Pharm. Res. 2017, 40, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekcec, A.; Yigitkanli, K.; Jung, J.E.; Pallast, S.; Xing, C.; Antipenko, A.; Minchenko, M.; Nikolov, D.B.; Holman, T.R.; Lo, E.H.; et al. Following experimental stroke, the recovering brain is vulnerable to lipoxygenase-dependent semaphorin signaling. FASEB J. 2013, 27, 437–445. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kim, J.H.; Lee, D.K.; Lee, K.S.; Won, M.H.; Jeoung, D.; Lee, H.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Carbon Monoxide Potentiation of L-Type Ca2+ Channel Activity Increases HIF-1alpha-Independent VEGF Expression via an AMPKalpha/SIRT1-Mediated PGC-1alpha/ERRalpha Axis. Antioxid. Redox Signal. 2017, 27, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Park, J.H.; Yun, J.A.; Cha, J.H.; Kim, Y.; Won, M.H.; Kim, K.W.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Heme oxygenase metabolites improve astrocytic mitochondrial function via a Ca2+-dependent HIF-1alpha/ERRalpha circuit. PLoS ONE 2018, 13, e0202039. [Google Scholar]
- Choi, Y.K.; Kim, C.K.; Lee, H.; Jeoung, D.; Ha, K.S.; Kwon, Y.G.; Kim, K.W.; Kim, Y.M. Carbon monoxide promotes VEGF expression by increasing HIF-1alpha protein level via two distinct mechanisms, translational activation and stabilization of HIF-1alpha protein. J. Biol. Chem. 2010, 285, 32116–32125. [Google Scholar] [CrossRef] [PubMed]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Dore, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dore, S.; Sampei, K.; Goto, S.; Alkayed, N.J.; Guastella, D.; Blackshaw, S.; Gallagher, M.; Traystman, R.J.; Hurn, P.D.; Koehler, R.C.; et al. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol. Med. 1999, 5, 656–663. [Google Scholar] [CrossRef]
- Chen, K.; Gunter, K.; Maines, M.D. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem. 2000, 75, 304–313. [Google Scholar] [CrossRef]
- Wan, P.; Su, W.; Zhang, Y.; Li, Z.; Deng, C.; Zhuo, Y. Trimetazidine protects retinal ganglion cells from acute glaucoma via the Nrf2/Ho-1 pathway. Clin. Sci. (Lond) 2017, 131, 2363–2375. [Google Scholar] [CrossRef] [Green Version]
- Catino, S.; Paciello, F.; Miceli, F.; Rolesi, R.; Troiani, D.; Calabrese, V.; Santangelo, R.; Mancuso, C. Ferulic Acid Regulates the Nrf2/Heme Oxygenase-1 System and Counteracts Trimethyltin-Induced Neuronal Damage in the Human Neuroblastoma Cell Line SH-SY5Y. Front. Pharmacol. 2015, 6, 305. [Google Scholar] [CrossRef]
- Song, W.; Cressatti, M.; Zukor, H.; Liberman, A.; Galindez, C.; Schipper, H.M. Parkinsonian features in aging GFAP.HMOX1 transgenic mice overexpressing human HO-1 in the astroglial compartment. Neurobiol. Aging 2017, 58, 163–179. [Google Scholar] [CrossRef]
- Li, M.H.; Jang, J.H.; Na, H.K.; Cha, Y.N.; Surh, Y.J. Carbon monoxide produced by heme oxygenase-1 in response to nitrosative stress induces expression of glutamate-cysteine ligase in PC12 cells via activation of phosphatidylinositol 3-kinase and Nrf2 signaling. J. Biol. Chem. 2007, 282, 28577–28586. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Sawle, P.; Homer-Vanniasinkam, S.; Green, C.J.; Foresti, R.; Motterlini, R. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Crit. Care Med. 2012, 40, 544–552. [Google Scholar] [CrossRef]
- Lin, C.C.; Yang, C.C.; Hsiao, L.D.; Chen, S.Y.; Yang, C.M. Heme Oxygenase-1 Induction by Carbon Monoxide Releasing Molecule-3 Suppresses Interleukin-1beta-Mediated Neuroinflammation. Front. Mol. Neurosci. 2017, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Panahian, N.; Yoshiura, M.; Maines, M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J. Neurochem. 1999, 72, 1187–1203. [Google Scholar] [CrossRef]
- Shi, H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr. Med. Chem. 2009, 16, 4593–4600. [Google Scholar] [CrossRef]
- Esposito, E.; Hayakawa, K.; Ahn, B.J.; Chan, S.J.; Xing, C.; Liang, A.C.; Kim, K.W.; Arai, K.; Lo, E.H. Effects of ischemic post-conditioning on neuronal VEGF regulation and microglial polarization in a rat model of focal cerebral ischemia. J. Neurochem. 2018, 146, 160–172. [Google Scholar] [CrossRef]
- Goven, D.; Boutten, A.; Lecon-Malas, V.; Marchal-Somme, J.; Soler, P.; Boczkowski, J.; Bonay, M. Induction of heme oxygenase-1, biliverdin reductase and H-ferritin in lung macrophage in smokers with primary spontaneous pneumothorax: Role of HIF-1alpha. PLoS ONE 2010, 5, e10886. [Google Scholar] [CrossRef]
- Panahian, N.; Huang, T.; Maines, M.D. Enhanced neuronal expression of the oxidoreductase--biliverdin reductase--after permanent focal cerebral ischemia. Brain Res. 1999, 850, 1–13. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, J.H.; Baek, Y.Y.; Won, M.H.; Jeoung, D.; Lee, H.; Ha, K.S.; Kwon, Y.G.; Kim, Y.M. Carbon monoxide stimulates astrocytic mitochondrial biogenesis via L-type Ca2+ channel-mediated PGC-1alpha/ERRalpha activation. Biochem. Biophys. Res. Commun. 2016, 479, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Che, X.; Fang, Y.; Si, X.; Wang, J.; Hu, X.; Reis, C.; Chen, S. The Role of Gaseous Molecules in Traumatic Brain Injury: An Updated Review. Front. Neurosci. 2018, 12, 392. [Google Scholar] [CrossRef]
- Mancuso, C. Bilirubin and brain: A pharmacological approach. Neuropharmacology 2017, 118, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Capone, C.; Ranieri, S.C.; Fusco, S.; Calabrese, V.; Eboli, M.L.; Preziosi, P.; Galeotti, T.; Pani, G. Bilirubin as an endogenous modulator of neurotrophin redox signaling. J. Neurosci. Res. 2008, 86, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Fukushima, H.; Mukawa, T.; Toyoda, H.; Wu, L.J.; Zhao, M.G.; Xu, H.; Shang, Y.; Endoh, K.; Iwamoto, T.; et al. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J. Neurosci. 2011, 31, 8786–8802. [Google Scholar] [CrossRef] [PubMed]
- Biojone, C.; Casarotto, P.C.; Joca, S.R.; Castren, E. Interplay Between Nitric Oxide and Brain-Derived Neurotrophic Factor in Neuronal Plasticity. CNS Neurol. Disord. Drug Targets 2015, 14, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Trombino, S.; Cassano, R.; Sgambato, A.; De Paola, B.; Di Stasio, E.; Picci, N.; Preziosi, P.; Mancuso, C. Characterization of the S-denitrosylating activity of bilirubin. J Cell Mol. Med. 2009, 13, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Ewing, J.F.; Weber, C.M.; Maines, M.D. Biliverdin reductase is heat resistant and coexpressed with constitutive and heat shock forms of heme oxygenase in brain. J. Neurochem. 1993, 61, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Park, D.Y.; Lee, J.; Kim, J.; Kim, K.; Hong, S.; Han, S.; Kubota, Y.; Augustin, H.G.; Ding, L.; Kim, J.W.; et al. Plastic roles of pericytes in the blood-retinal barrier. Nat. Commun. 2017, 8, 15296. [Google Scholar] [CrossRef]
- Yoshioka, T.; Nagaoka, T.; Song, Y.; Yokota, H.; Tani, T.; Yoshida, A. Role of neuronal nitric oxide synthase in regulating retinal blood flow during flicker-induced hyperemia in cats. Investig. Ophthalmol. Vis. Sci. 2015, 56, 3113–3120. [Google Scholar] [CrossRef]
- Arai-Gaun, S.; Katai, N.; Kikuchi, T.; Kurokawa, T.; Ohta, K.; Yoshimura, N. Heme oxygenase-1 induced in muller cells plays a protective role in retinal ischemia-reperfusion injury in rats. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4226–4232. [Google Scholar] [CrossRef]
- Ingi, T.; Cheng, J.; Ronnett, G.V. Carbon monoxide: An endogenous modulator of the nitric oxide-cyclic GMP signaling system. Neuron 1996, 16, 835–842. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Qiu, S.; Wang, Y.V.; Park, S.J.H.; Mohns, E.J.; Mehta, B.; Liu, X.; Chang, B.; Zenisek, D.; Crair, M.C.; et al. Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature 2018, 560, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Dong, S.; Zhu, M.; Sherry, D.M.; Wang, C.; You, Z.; Haigh, J.J.; Le, Y.Z. Muller Glia Are a Major Cellular Source of Survival Signals for Retinal Neurons in Diabetes. Diabetes 2015, 64, 3554–3563. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Zhang, X.; Zhu, H. Inverse Relationship Between Serum Bilirubin Levels and Diabetic Foot in Chinese Patients with Type 2 Diabetes Mellitus. Med. Sci. Monit. 2017, 23, 5916–5923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, F.; Deng, X.; Yu, Y.; Chen, X.; Shen, L.; Zhong, X.; Qiu, W.; Jiang, Y.; Zhang, J.; Hu, X. Serum bilirubin concentrations and multiple sclerosis. J. Clin. Neurosci. 2011, 18, 1355–1359. [Google Scholar] [CrossRef]
- Novotny, L.; Vitek, L. Inverse relationship between serum bilirubin and atherosclerosis in men: A meta-analysis of published studies. Exp. Biol. Med. (Maywood) 2003, 228, 568–571. [Google Scholar] [CrossRef]
- Kim, E.S.; Mo, E.Y.; Moon, S.D.; Han, J.H. Inverse association between serum bilirubin levels and arterial stiffness in Korean women with type 2 diabetes. PLoS ONE 2014, 9, e109251. [Google Scholar] [CrossRef] [PubMed]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARalpha Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Dore, S.; Ferris, C.D.; Tomita, T.; Sawa, A.; Wolosker, H.; Borchelt, D.R.; Iwatsubo, T.; Kim, S.H.; Thinakaran, G.; et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 2000, 28, 461–473. [Google Scholar] [CrossRef]
- Hettiarachchi, N.; Dallas, M.; Al-Owais, M.; Griffiths, H.; Hooper, N.; Scragg, J.; Boyle, J.; Peers, C. Heme oxygenase-1 protects against Alzheimer’s amyloid-beta(1-42)-induced toxicity via carbon monoxide production. Cell Death Dis. 2014, 5, e1569. [Google Scholar] [CrossRef] [PubMed]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dattilo, S.; Mancuso, C.; Koverech, G.; Di Mauro, P.; Ontario, M.L.; Petralia, C.C.; Petralia, A.; Maiolino, L.; Serra, A.; Calabrese, E.J.; et al. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing 2015, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Cassano, T.; Arena, A.; Tramutola, A.; Lavecchia, M.A.; Coccia, R.; Butterfield, D.A.; Perluigi, M. Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic. Biol. Med. 2016, 91, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Sultana, R.; Coccia, R.; Mancuso, C.; Perluigi, M.; Butterfield, D.A. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic. Biol. Med. 2012, 52, 2292–2301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palozza, P.; Serini, S.; Curro, D.; Calviello, G.; Igarashi, K.; Mancuso, C. beta-Carotene and cigarette smoke condensate regulate heme oxygenase-1 and its repressor factor Bach1: Relationship with cell growth. Antioxid. Redox Signal. 2006, 8, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S.; Nakayama, M.; Kitamuro, T.; Udono-Fujimori, R.; Takahashi, K. Repression of heme oxygenase-1 expression as a defense strategy in humans. Exp. Biol. Med. (Maywood) 2003, 228, 472–473. [Google Scholar] [CrossRef]
- Nakayama, M.; Takahashi, K.; Kitamuro, T.; Yasumoto, K.; Katayose, D.; Shirato, K.; Fujii-Kuriyama, Y.; Shibahara, S. Repression of heme oxygenase-1 by hypoxia in vascular endothelial cells. Biochem. Biophys. Res. Commun. 2000, 271, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, K.; Kimura, T.; Yoshitake, J.; Akaike, T.; Shono, M.; Takamatsu, J.; Katsuragi, S.; Kitamura, T.; Miyakawa, T. Possible assessment for antioxidant capacity in Alzheimer’s disease by measuring lymphocyte heme oxygenase-1 expression with real-time RT-PCR. Ann. N. Y. Acad. Sci. 2002, 977, 173–178. [Google Scholar] [CrossRef]
- Barone, E.; Di Domenico, F.; Mancuso, C.; Butterfield, D.A. The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol. Dis. 2014, 62, 144–159. [Google Scholar] [CrossRef]
- Chen, W.; Maghzal, G.J.; Ayer, A.; Suarna, C.; Dunn, L.L.; Stocker, R. Absence of the biliverdin reductase-a gene is associated with increased endogenous oxidative stress. Free Radic. Biol. Med. 2018, 115, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Butterfield, D.A. Statins more than cholesterol lowering agents in Alzheimer disease: Their pleiotropic functions as potential therapeutic targets. Biochem. Pharmacol. 2014, 88, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, E.; Mancuso, C.; Di Domenico, F.; Sultana, R.; Murphy, M.P.; Head, E.; Butterfield, D.A. Biliverdin reductase-A: A novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J. Neurochem. 2012, 120, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Barone, E.; Di Domenico, F.; Cenini, G.; Sultana, R.; Murphy, M.P.; Mancuso, C.; Head, E. Atorvastatin treatment in a dog preclinical model of Alzheimer’s disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int. J. Neuropsychopharmacol. 2012, 15, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Peltier, J.; O’Neill, A.; Schaffer, D.V. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev. Neurobiol. 2007, 67, 1348–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Lee, S.; Shrestha, P.; Kim, J.; Park, J.A.; Ko, Y.; Ban, Y.H.; Park, D.Y.; Ha, S.J.; Koh, G.Y.; et al. AMIGO2, a novel membrane anchor of PDK1, controls cell survival and angiogenesis via Akt activation. J. Cell Biol. 2015, 211, 619–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na, H.J.; Hwang, J.Y.; Lee, K.S.; Choi, Y.K.; Choe, J.; Kim, J.Y.; Moon, H.E.; Kim, K.W.; Koh, G.Y.; Lee, H.; et al. TRAIL negatively regulates VEGF-induced angiogenesis via caspase-8-mediated enzymatic and non-enzymatic functions. Angiogenesis 2014, 17, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Deramaudt, B.M.; Braunstein, S.; Remy, P.; Abraham, N.G. Gene transfer of human heme oxygenase into coronary endothelial cells potentially promotes angiogenesis. J Cell. Biochem. 1998, 68, 121–127. [Google Scholar] [CrossRef]
- Suzuki, M.; Iso-o, N.; Takeshita, S.; Tsukamoto, K.; Mori, I.; Sato, T.; Ohno, M.; Nagai, R.; Ishizaka, N. Facilitated angiogenesis induced by heme oxygenase-1 gene transfer in a rat model of hindlimb ischemia. Biochem. Biophys. Res. Commun. 2003, 302, 138–143. [Google Scholar] [CrossRef]
- Ikeda, Y.; Hamano, H.; Satoh, A.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Ishizawa, K.; Aihara, K.; Tsuchiya, K.; Tamaki, T. Bilirubin exerts pro-angiogenic property through Akt-eNOS-dependent pathway. Hypertens Res. 2015, 38, 733–740. [Google Scholar] [CrossRef]
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Zhu, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Greenberg, D.A. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, S.Y.; Liou, H.C.; Fu, W.M. The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology 2010, 58, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Kim, E.; Ratan, R.; Lee, F.S.; Cho, S. Genetic variant of BDNF (Val66Met) polymorphism attenuates stroke-induced angiogenic responses by enhancing anti-angiogenic mediator CD36 expression. J. Neurosci. 2011, 31, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.Y.; Liou, H.C.; Kang, K.H.; Wu, R.M.; Wen, C.C.; Fu, W.M. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wang, S.; Cai, J.; Rao, M.S.; Mattson, M.P. Nitric oxide acts in a positive feedback loop with BDNF to regulate neural progenitor cell proliferation and differentiation in the mammalian brain. Dev. Biol. 2003, 258, 319–333. [Google Scholar] [CrossRef] [Green Version]
- Qi, D.; Ouyang, C.; Wang, Y.; Zhang, S.; Ma, X.; Song, Y.; Yu, H.; Tang, J.; Fu, W.; Sheng, L.; et al. HO-1 attenuates hippocampal neurons injury via the activation of BDNF-TrkB-PI3K/Akt signaling pathway in stroke. Brain Res. 2014, 1577, 69–76. [Google Scholar] [CrossRef]
- Chen, T.; Yu, Y.; Tang, L.J.; Kong, L.; Zhang, C.H.; Chu, H.Y.; Yin, L.W.; Ma, H.Y. Neural stem cells over-expressing brain-derived neurotrophic factor promote neuronal survival and cytoskeletal protein expression in traumatic brain injury sites. Neural Regen. Res. 2017, 12, 433–439. [Google Scholar]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Li Calzi, S.; Lach, R.; Hock, T.D.; Chen, B.; Hill-Kapturczak, N.; et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase 1-dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef] [Green Version]
- Enzmann, V.; Lecaude, S.; Kruschinski, A.; Vater, A. CXCL12/SDF-1-Dependent Retinal Migration of Endogenous Bone Marrow-Derived Stem Cells Improves Visual Function after Pharmacologically Induced Retinal Degeneration. Stem Cell Rev. 2017, 13, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, S.; Smith, T.N.; Wang, N.; Chua, J.Y.; Westbroek, E.; Wang, K.; Guzman, R. BDNF Pretreatment of Human Embryonic-Derived Neural Stem Cells Improves Cell Survival and Functional Recovery After Transplantation in Hypoxic-Ischemic Stroke. Cell Transpl. 2015, 24, 2449–2461. [Google Scholar] [CrossRef] [PubMed]
- Ridiandries, A.; Tan, J.T.; Bursill, C.A. The Role of CC-Chemokines in the Regulation of Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1856. [Google Scholar] [CrossRef] [PubMed]
- Uede, T. Osteopontin, intrinsic tissue regulator of intractable inflammatory diseases. Pathol. Int. 2011, 61, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Dreyer-Andersen, N.; Almeida, A.S.; Jensen, P.; Kamand, M.; Okarmus, J.; Rosenberg, T.; Friis, S.D.; Martinez Serrano, A.; Blaabjerg, M.; Kristensen, B.W.; et al. Intermittent, low dose carbon monoxide exposure enhances survival and dopaminergic differentiation of human neural stem cells. PLoS ONE 2018, 13, e0191207. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Min, K.S.; Ryu, H.W.; Lee, H.J.; Kim, E.C. The role of heme oxygenase-1 in the proliferation and odontoblastic differentiation of human dental pulp cells. J. Endod. 2010, 36, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Raja, R.; Kale, S.; Thorat, D.; Soundararajan, G.; Lohite, K.; Mane, A.; Karnik, S.; Kundu, G.C. Hypoxia-driven osteopontin contributes to breast tumor growth through modulation of HIF1alpha-mediated VEGF-dependent angiogenesis. Oncogene 2014, 33, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Peng, L.; Fan, K.; Wang, H.; Wei, R.; Ji, G.; Cai, J.; Lu, B.; Li, B.; Zhang, D.; et al. Osteopontin induces angiogenesis through activation of PI3K/AKT and ERK1/2 in endothelial cells. Oncogene 2009, 28, 3412–3422. [Google Scholar] [CrossRef]
- Rogall, R.; Rabenstein, M.; Vay, S.; Bach, A.; Pikhovych, A.; Baermann, J.; Hoehn, M.; Couillard-Despres, S.; Fink, G.R.; Schroeter, M.; et al. Bioluminescence imaging visualizes osteopontin-induced neurogenesis and neuroblast migration in the mouse brain after stroke. Stem Cell Res. Ther. 2018, 9, 182. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef]

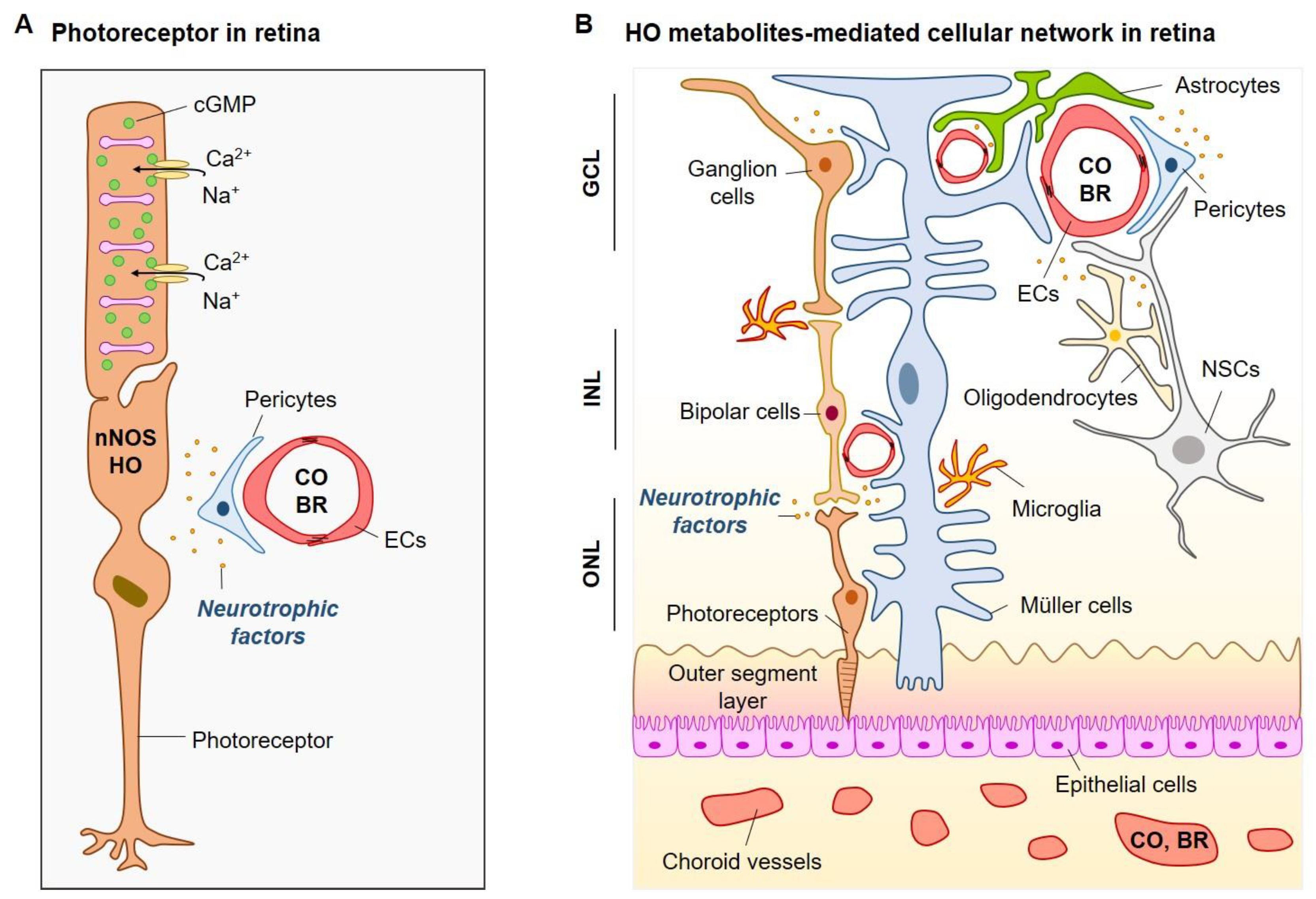

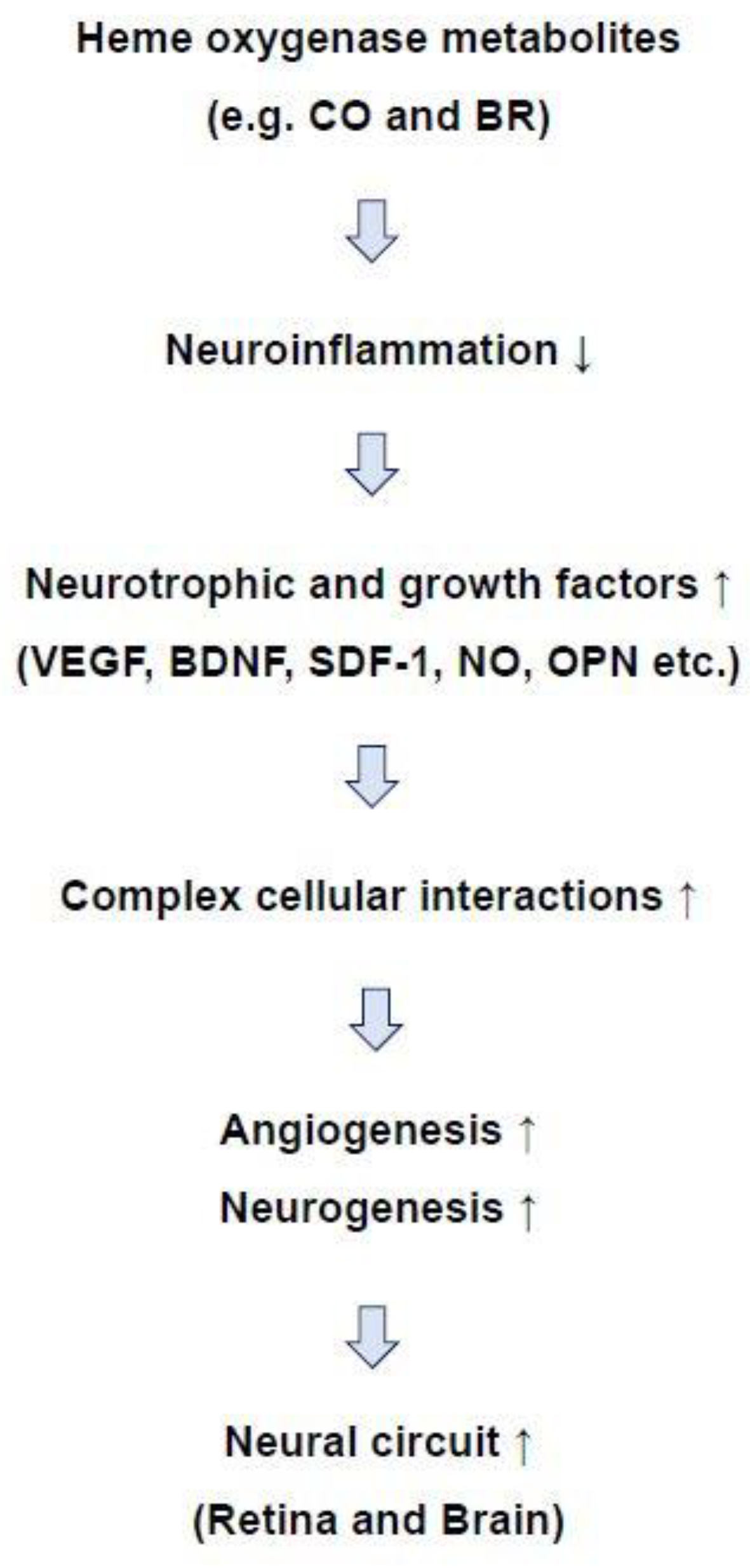
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.; Choi, Y.K. Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 78. https://doi.org/10.3390/ijms20010078
Lee H, Choi YK. Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases. International Journal of Molecular Sciences. 2019; 20(1):78. https://doi.org/10.3390/ijms20010078
Chicago/Turabian StyleLee, Huiju, and Yoon Kyung Choi. 2019. "Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases" International Journal of Molecular Sciences 20, no. 1: 78. https://doi.org/10.3390/ijms20010078
APA StyleLee, H., & Choi, Y. K. (2019). Regenerative Effects of Heme Oxygenase Metabolites on Neuroinflammatory Diseases. International Journal of Molecular Sciences, 20(1), 78. https://doi.org/10.3390/ijms20010078