2.1. Receptor Binding and Activation of CCR1 by Wild Type Chemokines
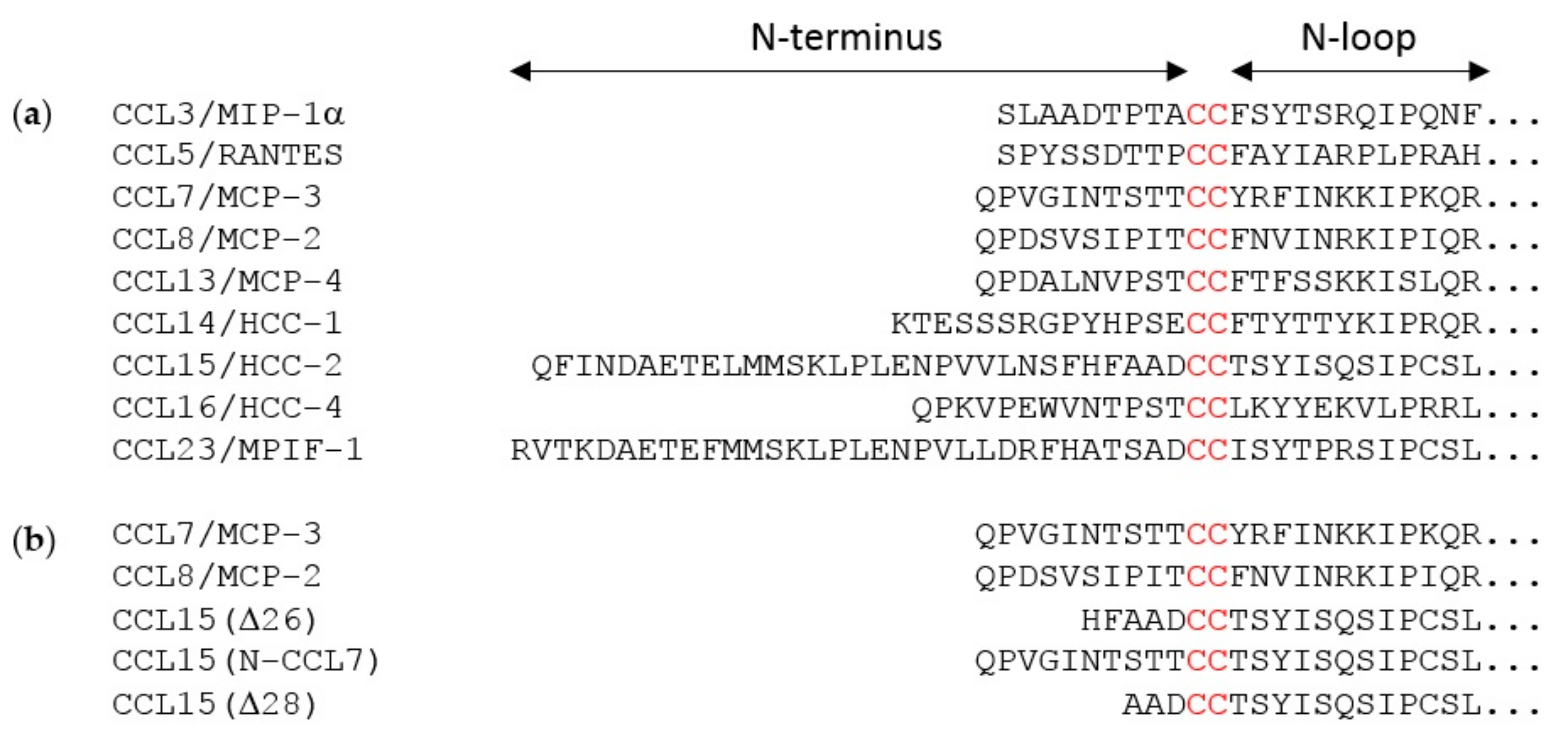
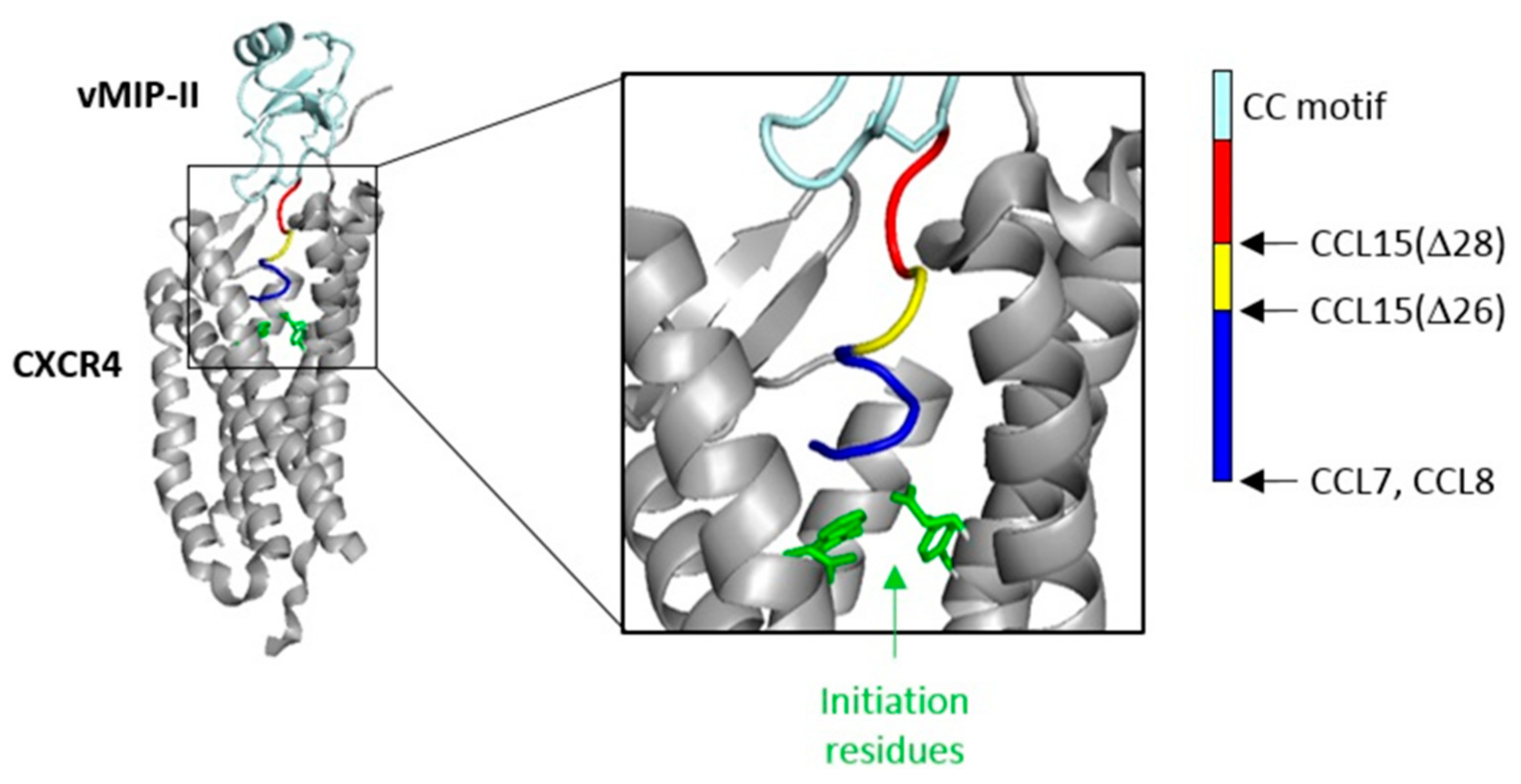
The flexible N-terminal regions, preceding the conserved CC or CXC motif, of most chemokines consist of ~8–10 amino acid residues. This enables the chemokine N-terminal region, in a largely extended conformation, to occupy the majority of space within the binding pocket defined by the TM helices of chemokine receptors. However, two of the cognate ligands of CCR1 (CCL15 and CCL23) have much longer N-terminal regions (31 and 32 residues, respectively). Full-length CCL15 is reported to have low potency of CCR1 activation, but a form of CCL15 with the first 26 residues removed, CCL15(Δ26), has much higher affinity and potency at CCR1, and further truncation results in moderate decreases in affinity and potency relative to CCL15(Δ26) [
29]. These shorter forms of CCL15 contain only five or fewer residues in their N-terminal regions, insufficient to occupy the TM binding pocket, raising the question of whether they achieve receptor activation by a different structural mechanism from other chemokines, potentially giving rise to biased agonism. To investigate this, we compared CCR1 binding and activation by three human chemokines: CCL7, CCL8, and CCL15(Δ26) (
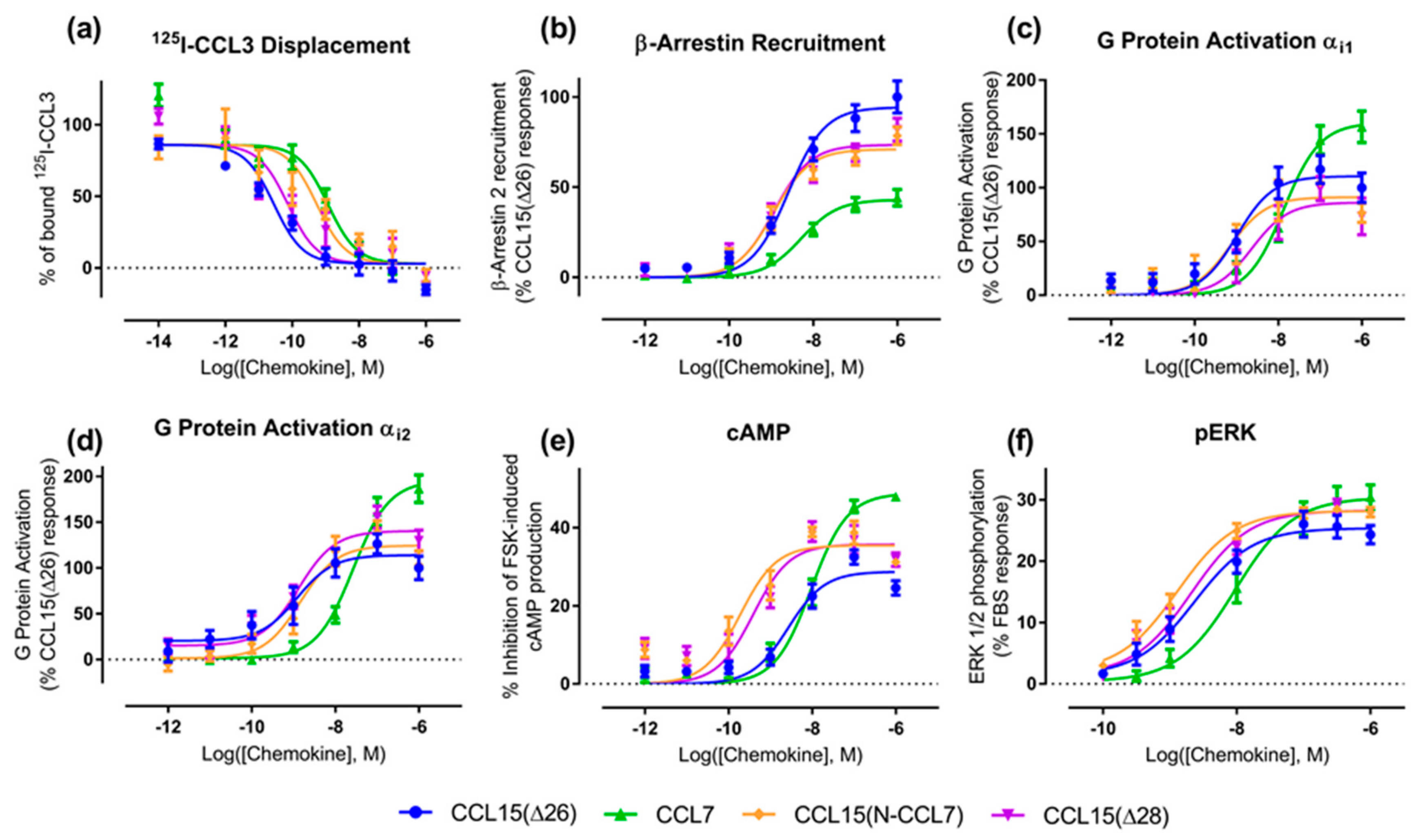
Figure 1b and
Figure 2).
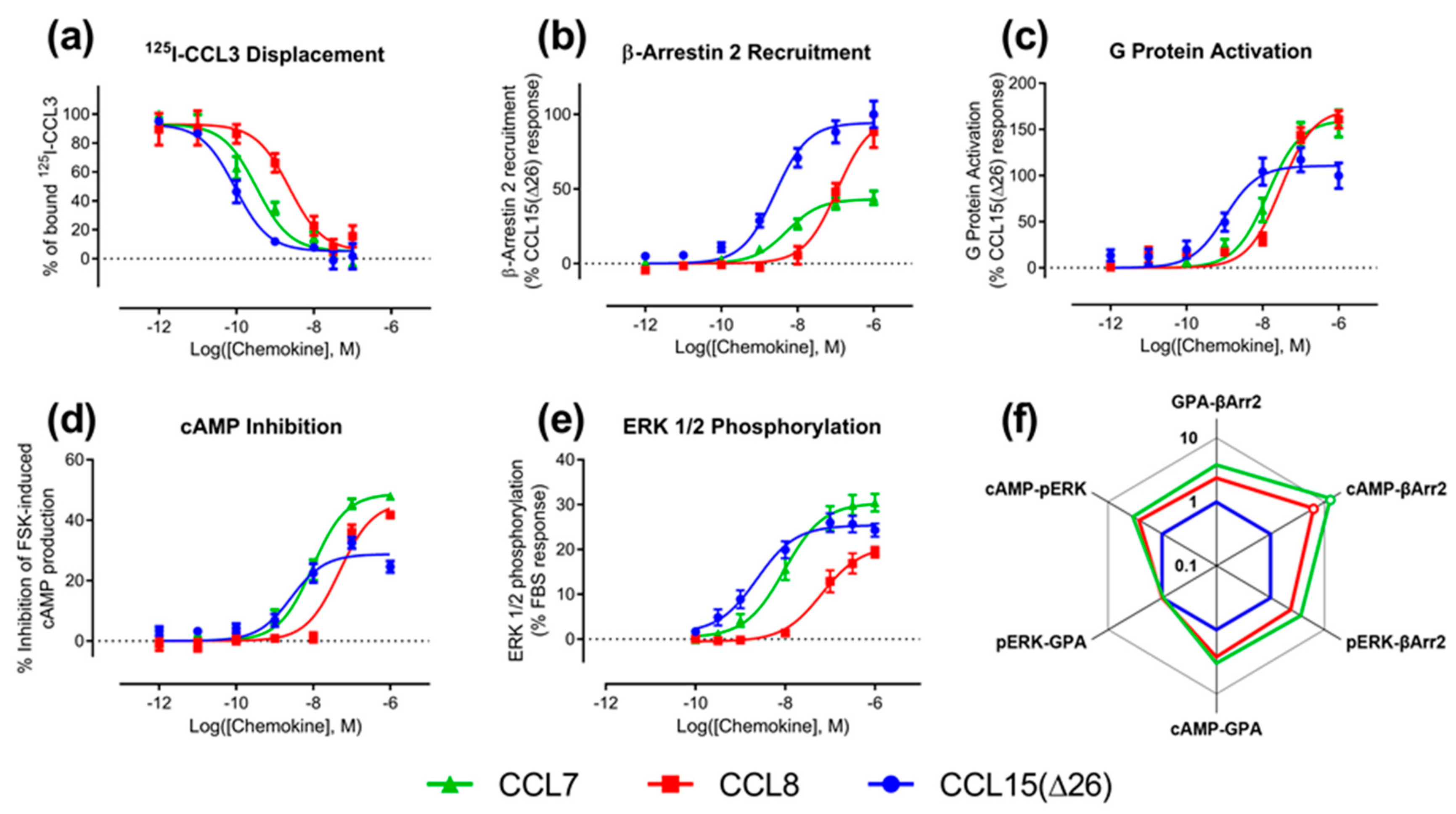
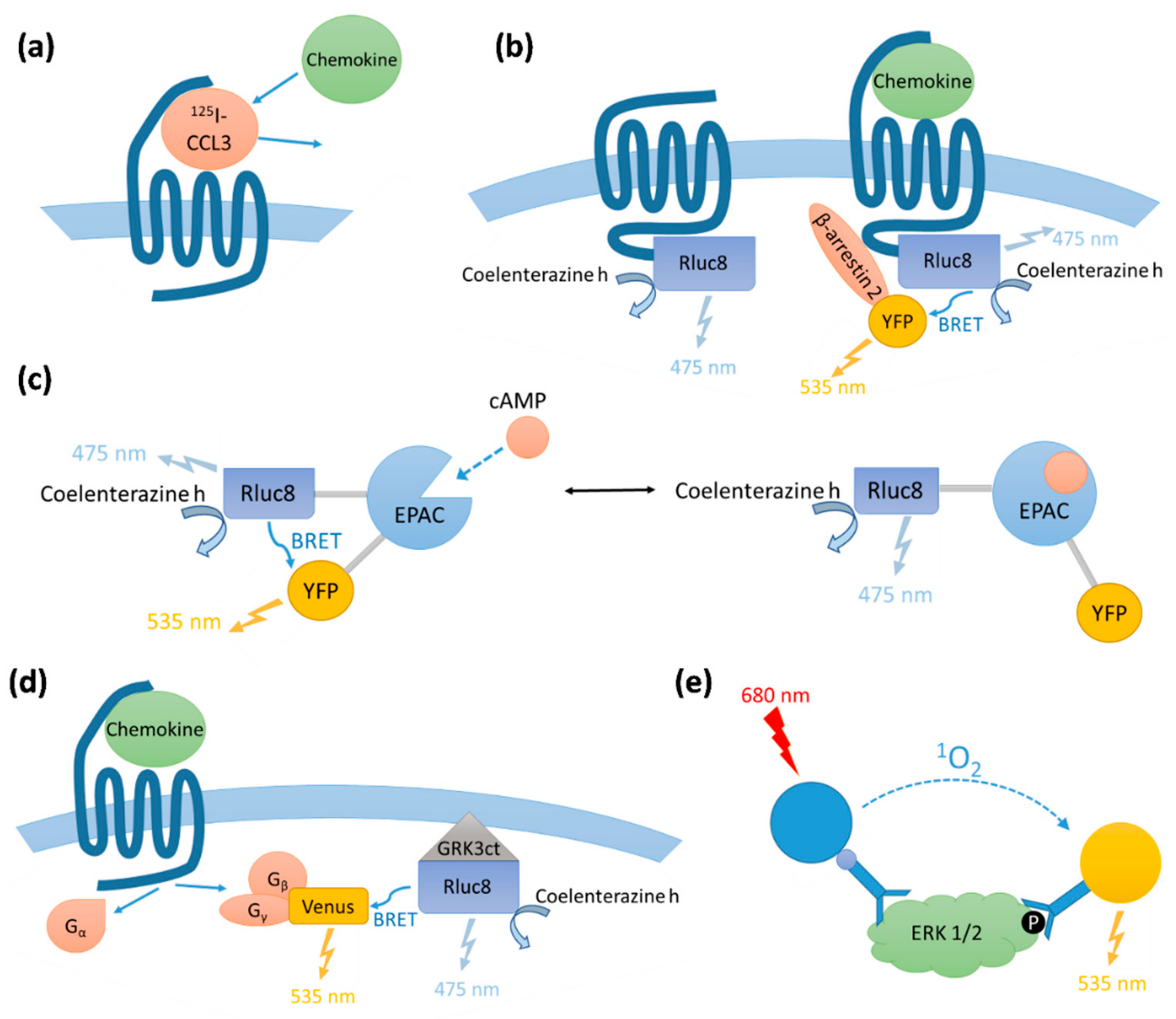
We determined the binding affinities of chemokines for CCR1 expressed in Flp-In T-REx human embryonic kidney (HEK) 293 cells, using a radioligand displacement assay (
Figure 2a and
Table 1). All three chemokines competed with
125I-labeled CCL3 for binding to CCR1 in a concentration-dependent manner. The data indicated that CCL15(Δ26) had the highest affinity, with half-maximal displacement at a concentration (
IC50) of 0.093 nM, whereas CCL7 and CCL8 had lower affinities with
IC50 values of 0.33 and 2.3 nM, respectively (
Table 1).
We compared the abilities of CCL7, CCL8, and CCL15(Δ26) to activate CCR1 using four different cell-based signaling assays, as measured 5–10 min after agonist stimulation (
Figure A1 and
Figure A2). Recruitment of βArr is a proximal (non-amplified) measure of receptor activation, whereas G
i1 protein activation, and the downstream signals of inhibition of cAMP production and phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), are all amplified to varying degrees. In all assays, all three chemokines stimulated concentration-dependent signaling via CCR1 (
Figure 2b–e and
Table 1). In the recruitment of βArr, CCL15(Δ26) and CCL7 had similar potencies (
pEC50), but CCL15(Δ26) exhibited a significantly higher maximal effect (
Emax) than CCL7 (
Figure 2b and
Table 1). In contrast, CCL8 had lower potency than the other two chemokines but its maximal effect was similar to that of CCL15(Δ26). In an initial indication of biased agonism, we observed that the order of potencies and maximal effects was not the same in all assays (
Table 2). For example, in all assays, CCL15(Δ26) displayed higher potency than CCL8, but the potency of CCL7 was similar to that of CCL8 in the G protein activation assay and similar to that of CCL15(Δ26) in the other three assays. In addition, CCL15(Δ26) displayed a significantly higher maximal effect than CCL7 in the βArr recruitment assay, but significantly lower maximal effect than CCL7 in both G protein activation and cAMP inhibition assays.
To detect and quantify biased agonism at CCR1, we analyzed the data for each concentration-response experiment using a derivation of the Black and Leff operational model of agonism [
30,
31]. This analysis yielded a “transduction coefficient”, log(τ/K
A), as a measure of intrinsic activity of an agonist at a given pathway, which was normalized relative to a reference ligand, CCL15(Δ26). Comparison of the normalized transduction coefficients across the different signaling pathways (
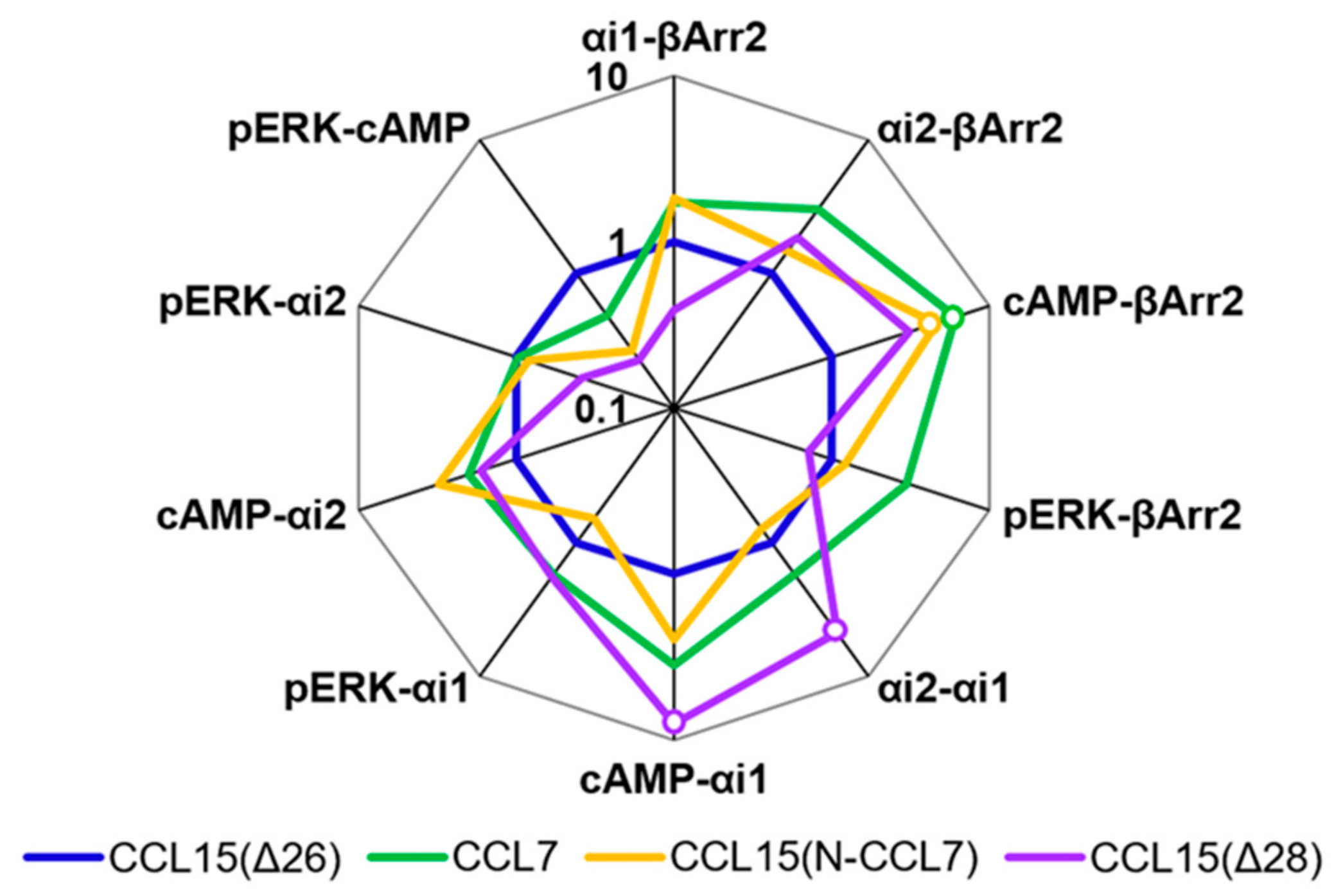
Figure 2f,
Table A1) revealed that CCL7 and CCL8 displayed biased agonism relative to CCL15(Δ26) with both chemokines showing bias towards cAMP inhibition and away from β-Arrestin 2 recruitment compared to CCL15(Δ26).
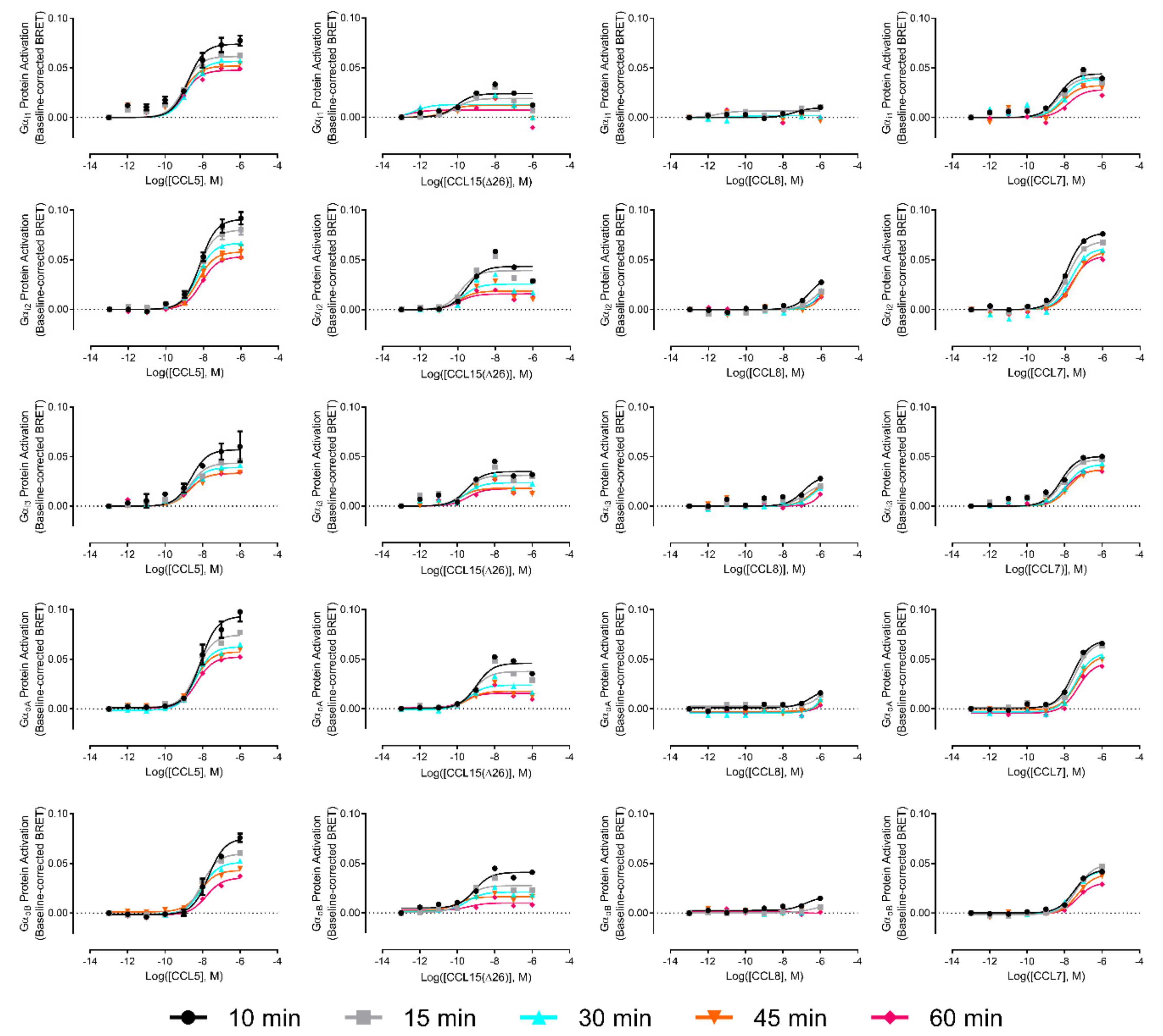
In addition to identifying significant biased activation of cAMP inhibition versus βArr pathways, the above analysis showed that, in comparison to CCL15(Δ26), CCL7 and CCL8 displayed a slight preference for stimulation of the cAMP inhibition response over both the ERK1/2 phosphorylation response and the G protein activation response. Although these comparisons did not reach statistical significance, they suggested that CCR1 might preferentially associate with certain G protein subtypes in a biased manner when activated by different ligands. Therefore, to investigate the possibilities of G protein subtype-coupling bias, we repeated the G protein activation assay using a set of five Gα subunits known to inhibit adenylyl cyclase and interact with chemokine receptors (i1, i2, i3, oA, and oB). Moreover, we recognized that the activation of different G proteins subtypes could potentially follow distinct time courses, thereby resulting in measurable bias when detected at certain time points. We thus used detection times of 10, 15, 30, 45, and 60 min after agonist stimulation. The concentration-response curves are presented in the
Appendix A (
Figure A3). Gα
i1 had the best coupling to CCR1, irrespective of the ligand used, although saturation was not reached for CCL8. CCL15(Δ26) was the most potent ligand and CCL5 was the most efficacious ligand, irrespective of the Gα protein subtype. The maximum response was observed 10 min after the addition of ligands and
Emax values decreased overtime. We did not observe any significant bias between activation of different Gα subtypes at any of the time points investigated.
2.2. Influence of Chemokine N-Terminal Sequence on Biased Agonism at CCR1
To test the hypothesis that the N-terminal region is the primary site influencing biased agonism at CCR1, we generated a chimeric chemokine consisting of CCL15 with the N-terminal region substituted by that of CCL7, named CCL15(N-CCL7) (
Figure 1b and
Figure A4a). CCL15(N-CCL7) bound to CCR1 with affinity indistinguishable from that of CCL7, which was significantly lower than the affinity of CCL15(Δ26), indicating that the N-terminal region of CCL15(Δ26) contributes to its increased CCR1 binding affinity (
Figure 3a,
Table 3). However, the effects of N-terminal substitution on CCR1 activation were different for the various functional assays (
Figure 3b–f,
Table 3). In both G protein activation (using Gα
i1 or Gα
i2) and ERK1/2 phosphorylation assays, the chimeric chemokine displayed concentration-response profiles similar to those of CCL15(Δ26), suggesting that the N-terminal regions of the two chemokines are equally capable of activating these pathways. In the βArr recruitment assay, CCL15(N-CCL7) displayed a maximal effect intermediate between the two parental chemokines, indicating that the N-terminal regions of the two chemokines contribute differentially to βArr recruitment. In the cAMP inhibition assay, CCL15(N-CCL7) displayed significantly higher potency than either CCL15(Δ26) or CCL7, suggesting that the N-terminus of CCL7 and other regions of CCL15(Δ26) were able to cooperatively and selectively stabilize the conformation of CCR1 giving rise to cAMP inhibition. Analysis of transduction coefficients for CCR1 activation indicated that, relative to CCL15(Δ26), CCL15(N-CCL7) is biased towards cAMP inhibition and away from βArr recruitment (
Figure 4,
Table A2). Although the bias profile of CCL15(N-CCL7) was similar to that of CCL7, the concentration-response curves indicate that the underlying causes of bias may be different for these two proteins.
2.3. Influence of CCL15 N-Terminal Length on Biased Agonism at CCR1
A previous study showed that truncation of the CCL15(Δ26) N-terminus by two additional residues, to CCL15(Δ28), resulted in a loss of affinity and a ~3-fold reduction of potency for CCR1 activation, as measured using an aequorin luminescence assay, which senses changes in intracellular Ca
2+ concentration [
29]. To investigate whether CCL15 truncation differentially influenced activation of various signaling pathways via CCR1, we expressed and purified CCL15(Δ28) (
Figure 1b and
Figure A4b) and compared its CCR1 binding and activation to those of CCL15(Δ26). In membrane preparations of cells expressing CCR1, CCL15(Δ28) and CCL15(Δ26) bound to CCR1 with similar affinities (
Figure 3a,
Table 3). However, N-terminal truncation resulted in different effects for the various signaling assays (
Figure 3b–f,
Table 3). In the ERK1/2 phosphorylation assay, the concentration-response profiles of CCL15(Δ28) and CCL15(Δ26) were indistinguishable. In the G protein activation assays, the two chemokines displayed similar potency but the maximal effect of CCL15(Δ28) was slightly lower than that of CCL15(Δ26) when Gα
i1 was used and slightly higher than that of CCL15(Δ26) when Gα
i2 was used, albeit not reaching statistical significance. In the βArr recruitment assay, CCL15(Δ28) displayed similar potency but slightly lower maximal effect than CCL15(Δ26), whereas in the cAMP inhibition assay, CCL15(Δ28) displayed significantly higher potency relative to CCL15(Δ26). Analysis of transduction coefficients showed that, relative to CCL15(Δ26), CCL15(Δ28) is biased towards Gα
i2 activation and away from Gα
i1 activation and towards cAMP inhibition and away from Gα
i1 activation (
Figure 4,
Table A2).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




