Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
1.1. The Main Functions of the Endoplasmic Reticulum in Eukaryotic Cells
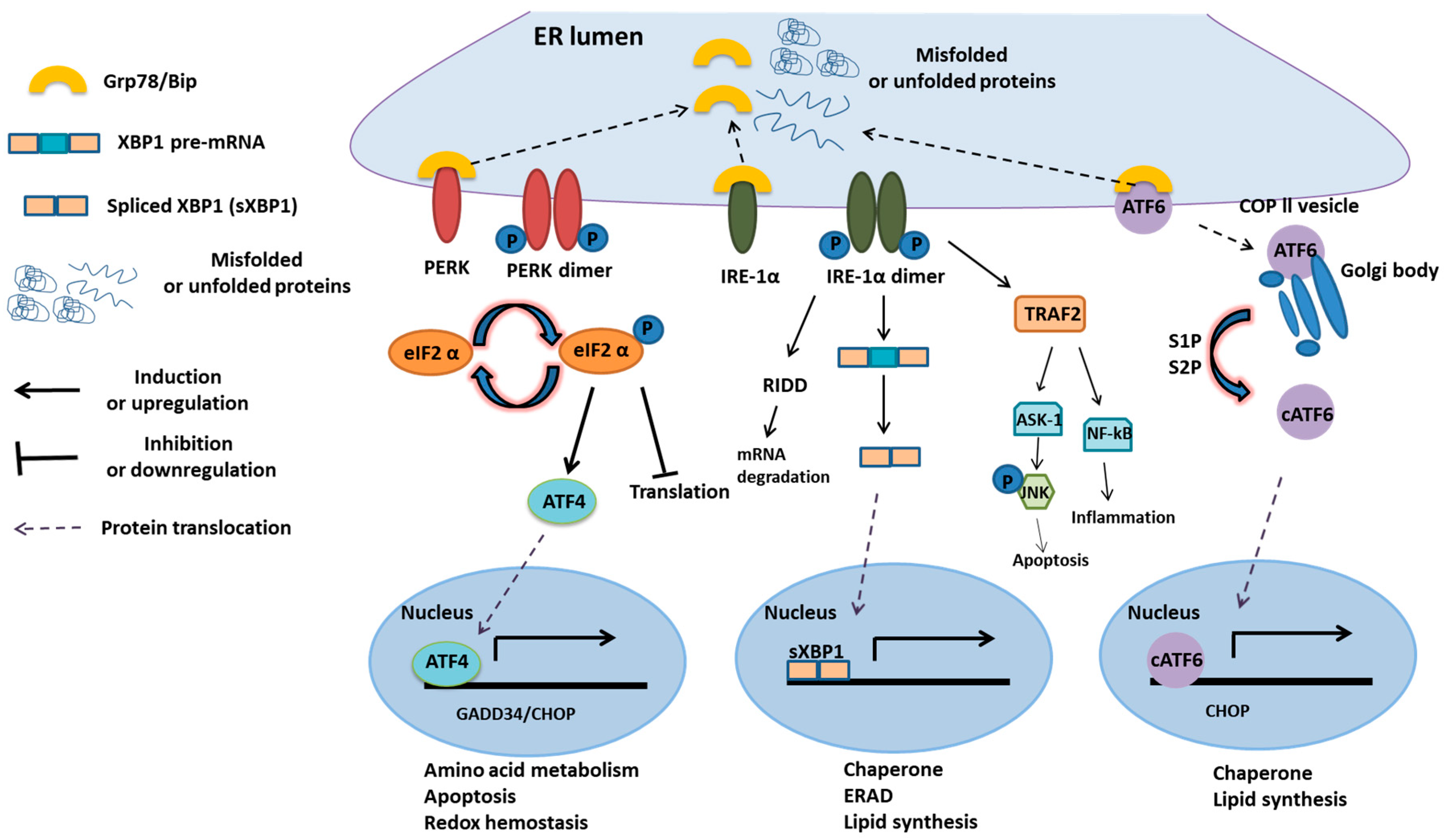
1.2. The Roles of UPR in Coping with Misfolded Proteins and Its Downstream Proteins
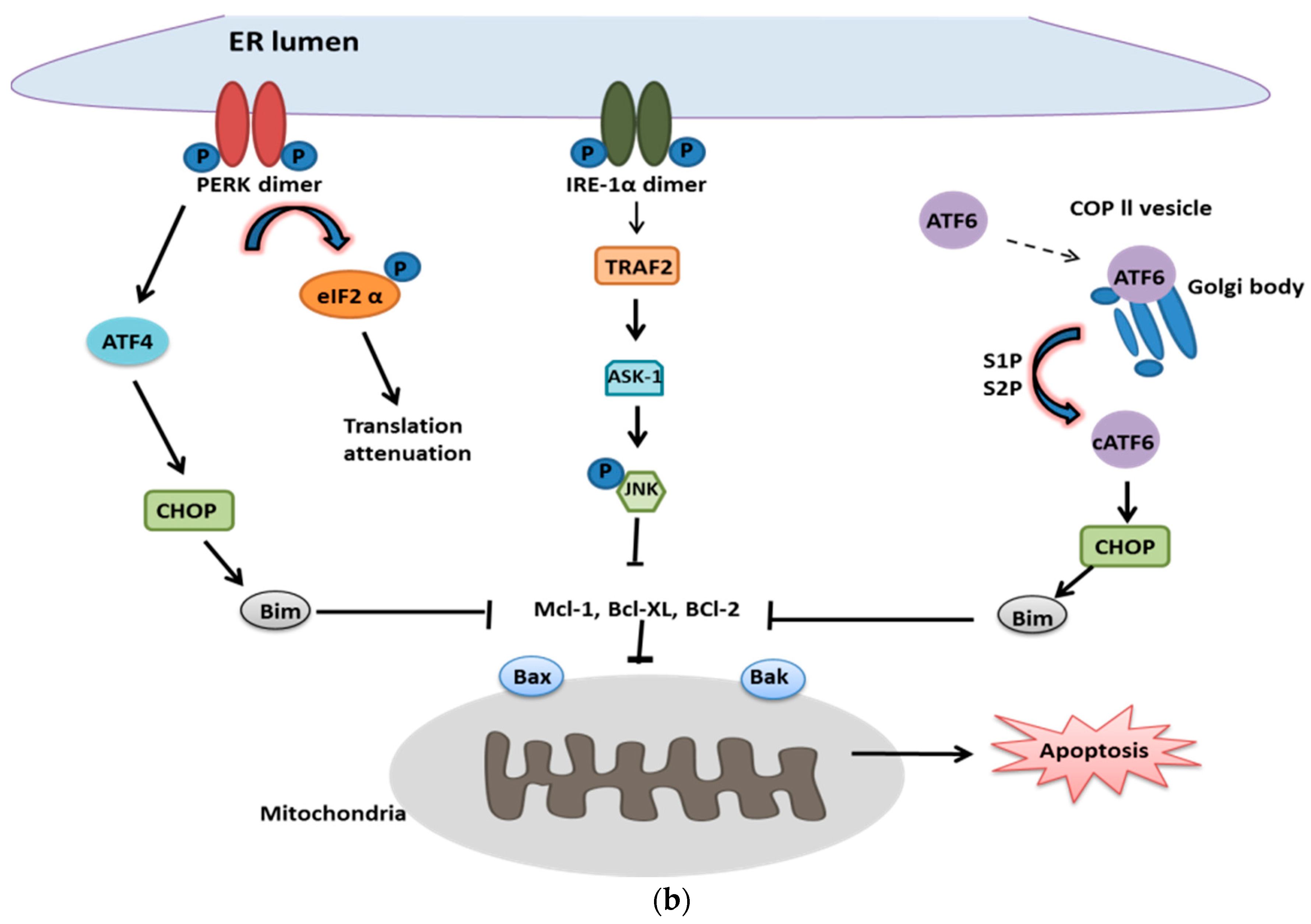
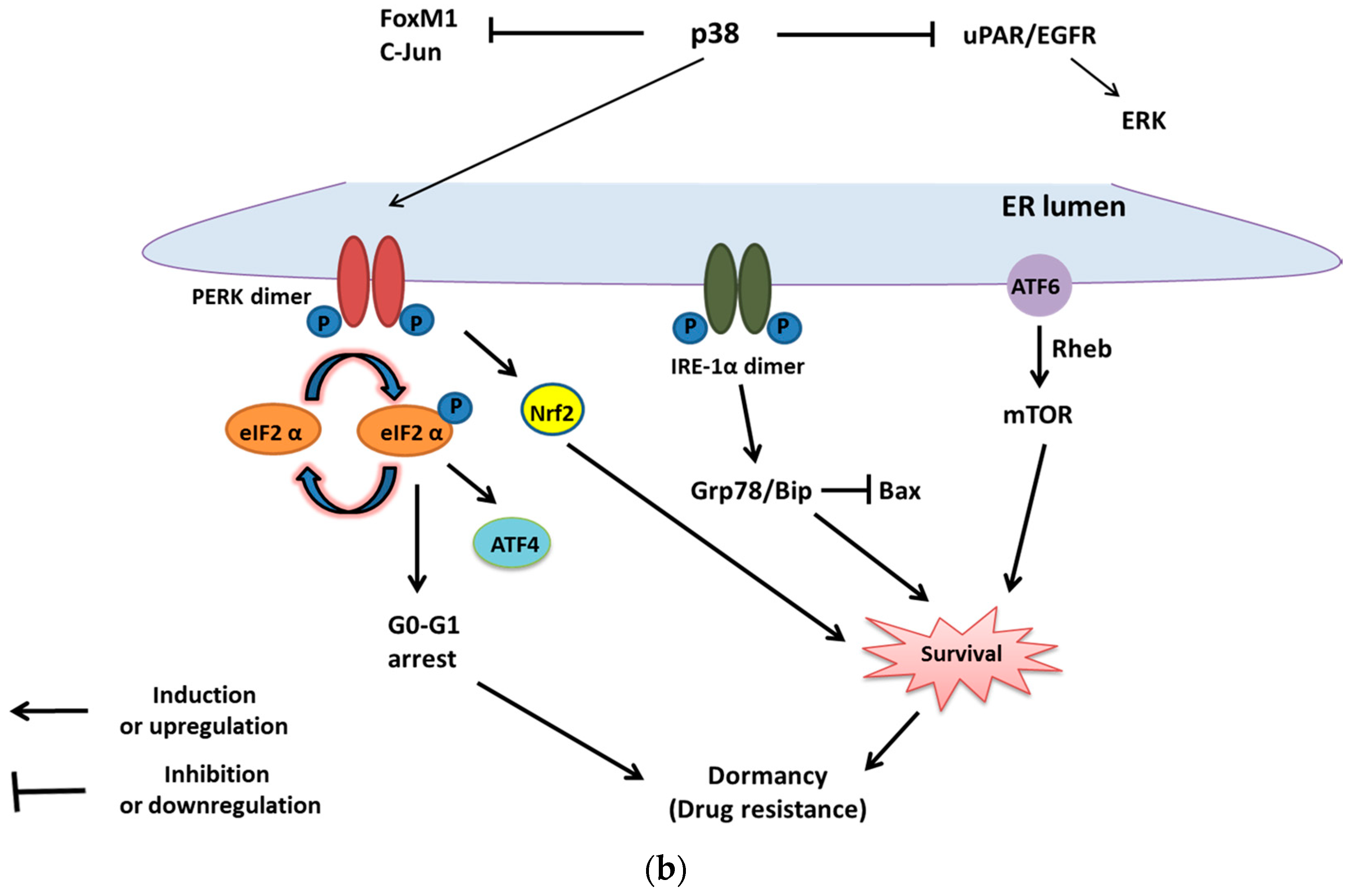
2. UPR and Cell Survival
2.1. UPR in Cell Survival
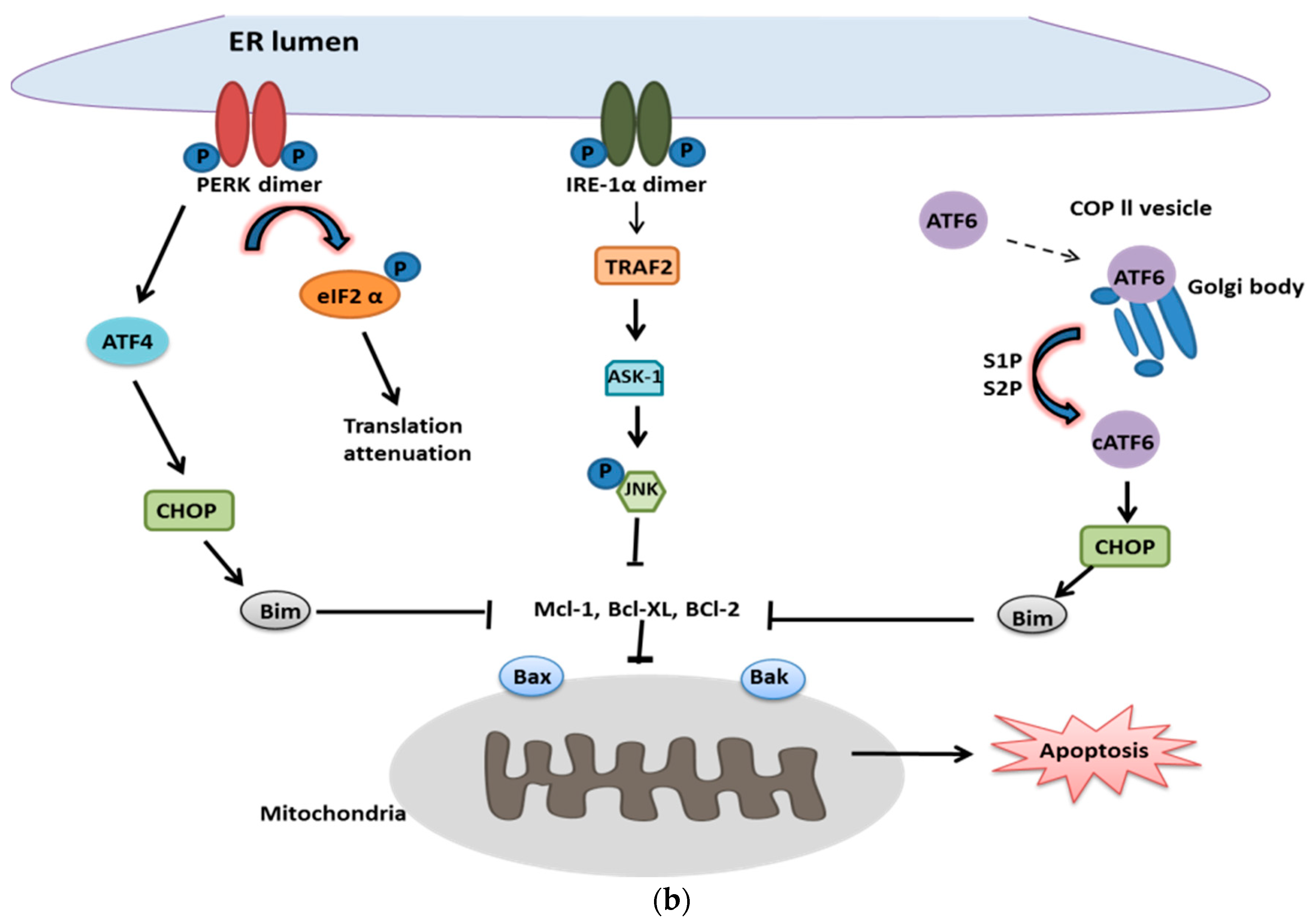
2.2. UPR in Cell Death
3. UPR and Tumor Dormancy
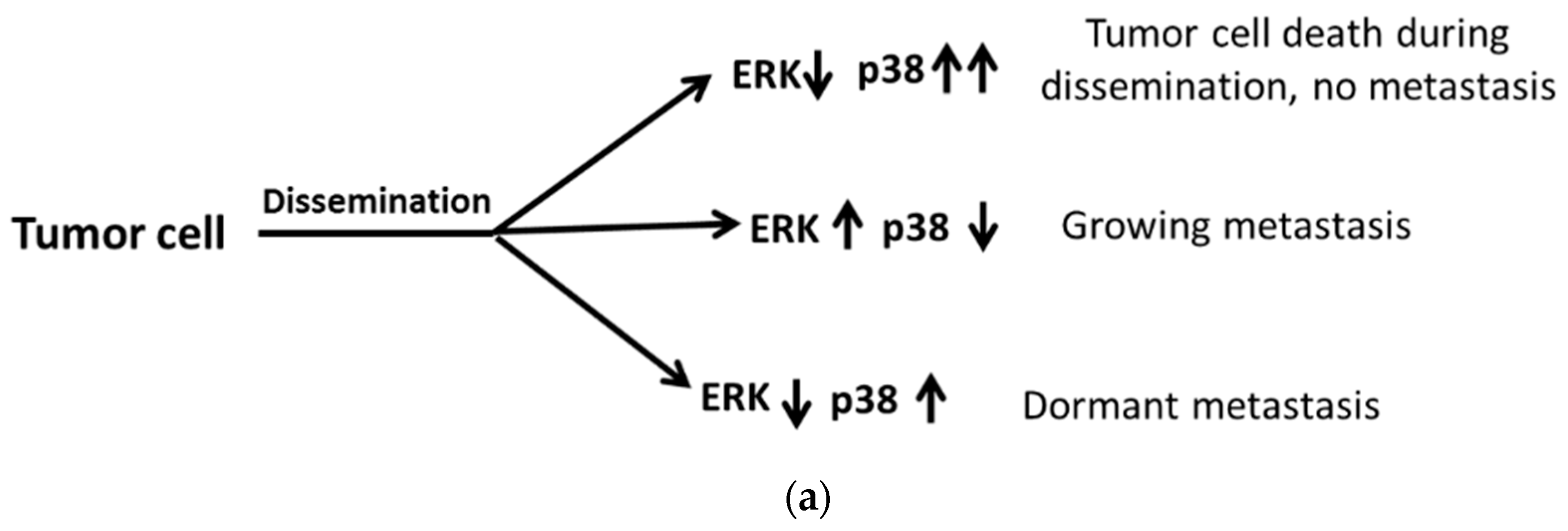
3.1. UPR-Induced Dormancy in Cancer Metastasis
3.2. The UPR-Induced Dormancy in Chemoresistance and Cell Survival
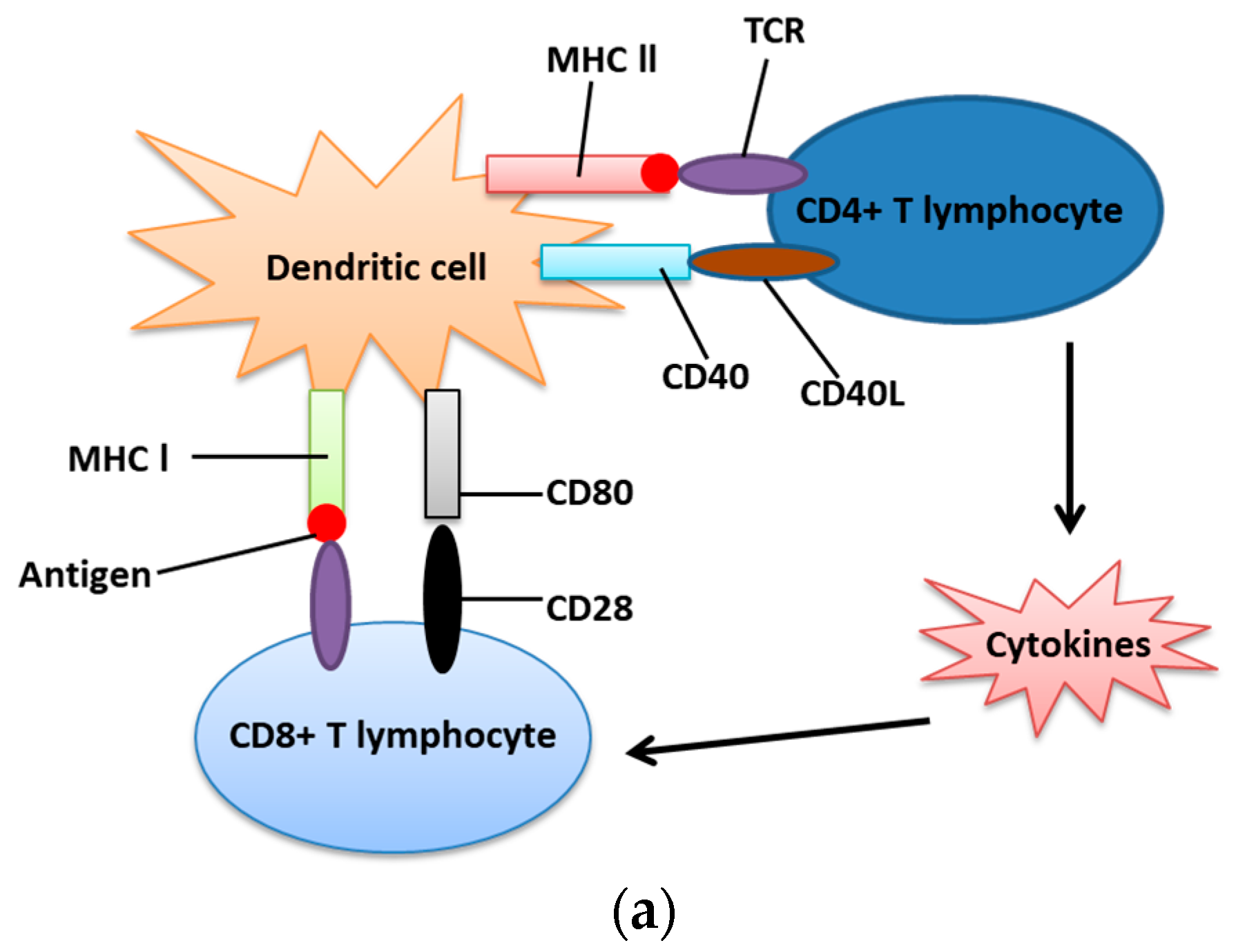
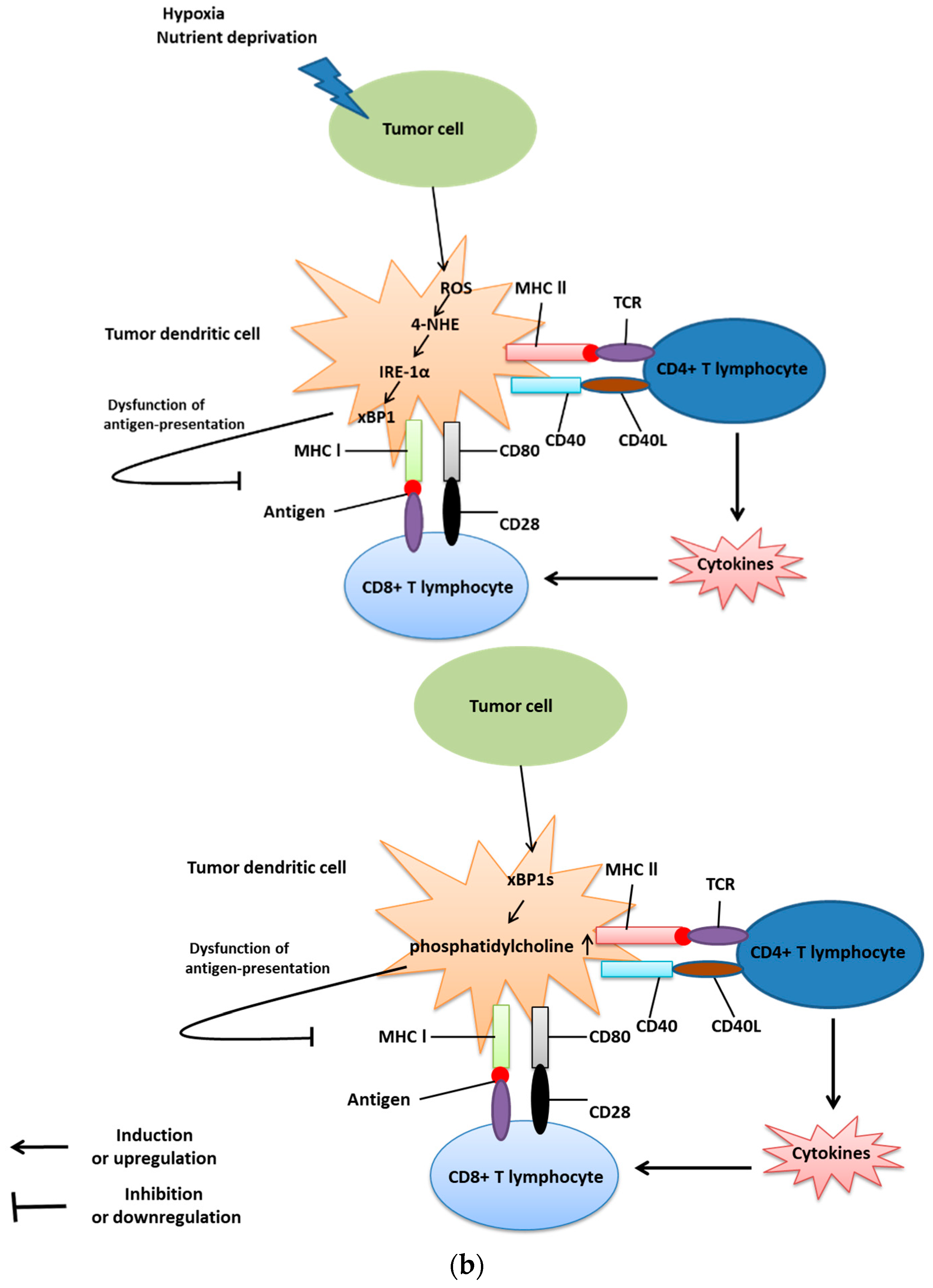
4. UPR and Immunosuppression in Cancer Cells
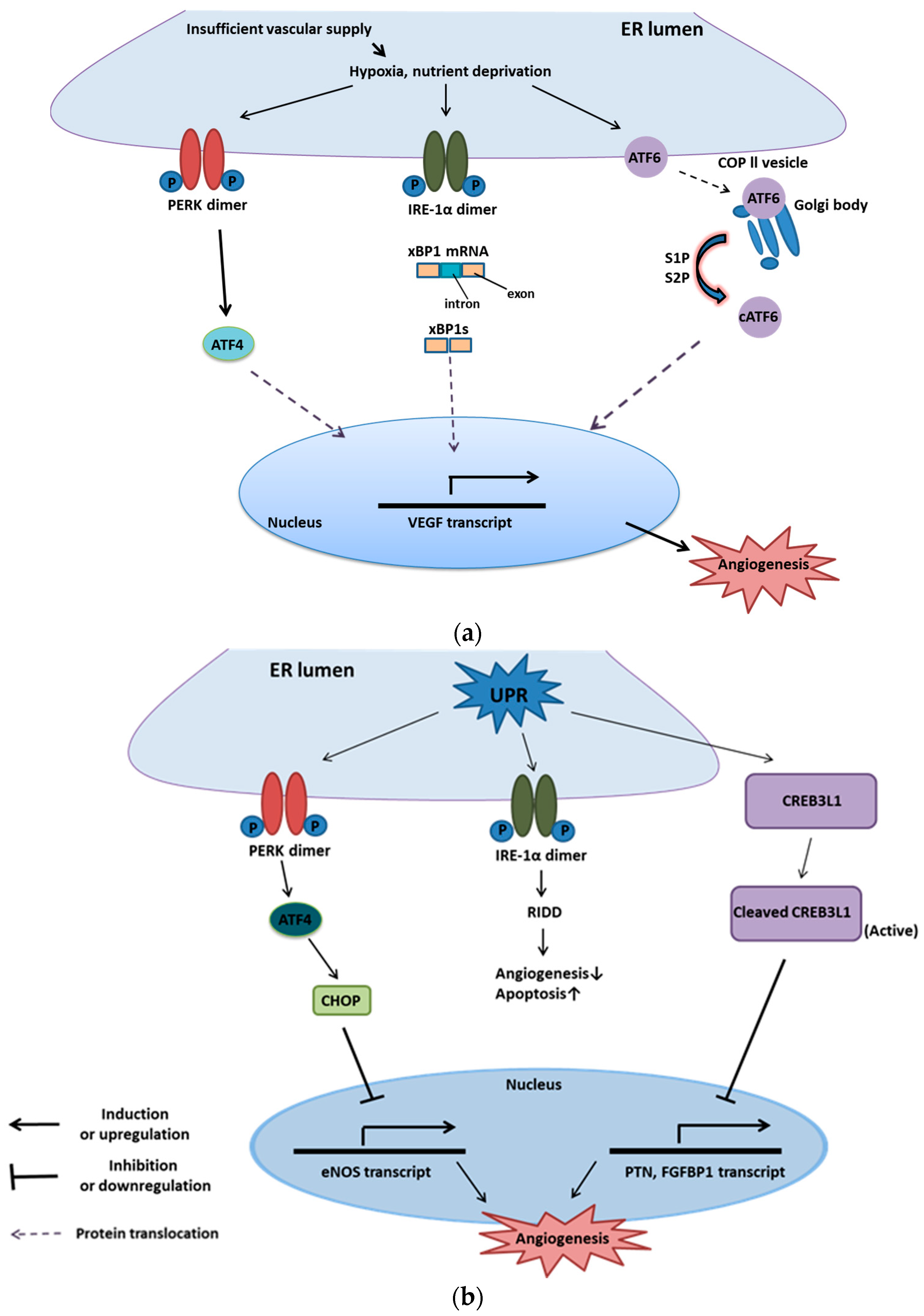
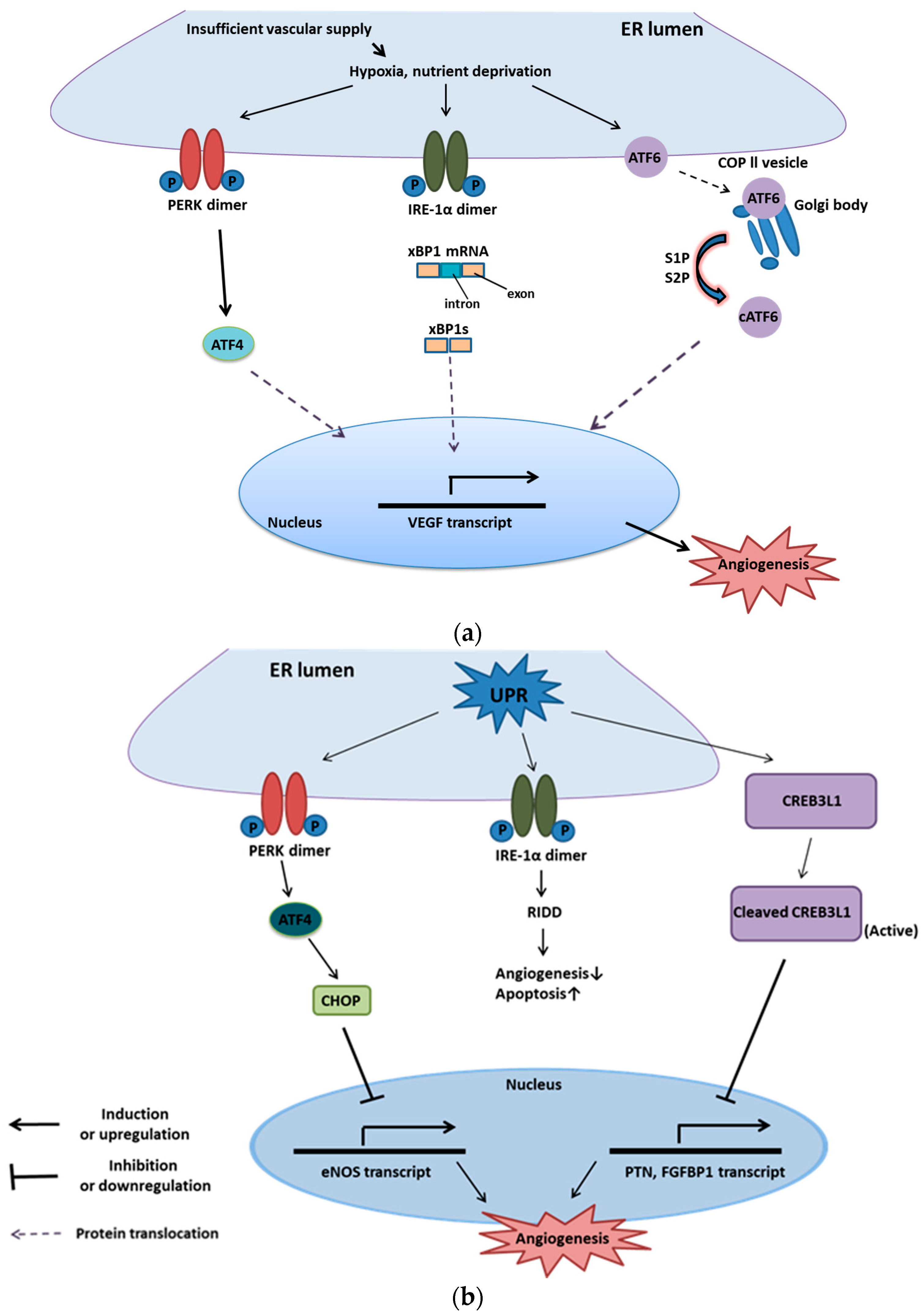
5. UPR and Angiogenesis
5.1. Introduction to Angiogenesis
5.2. Mechanism of Downstream UPR Regulating Angiogenesis
6. UPR and Metastasis
6.1. The Process of Metastasis
6.2. The Relationship between Metastasis and UPR
7. Promising Therapies that Inhibit Cancer Using Mediating UPR
7.1. Target for Grp78/Bip
7.2. Target for ATF6α
7.3. Target for IRE-1α
7.4. Target for PERK
7.5. Target for eIF2α
7.6. Target for ERAD
7.7. Target for Chaperones
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADCC | antibody-dependent cellular cytotoxicity |
| AEBSP | 4-(2-aminoethyl) benzene- sulfonyl fluoride |
| Akt | protein kinase B, PKB |
| ALS | amyotrophic lateral sclerosis |
| ASK-1 | apoptosis signal-regulating kinase 1 |
| ATF4 | activating transcription factor 4 |
| ATF6 | activating transcription factor 6 |
| CD40 | cluster of differentiation 40 |
| CDC | complement dependent cytotoxicity |
| CHOP | C/EBP homologous protein |
| CHOP10 | C/EBP homologous protein-10 |
| CREB3L1 | cyclic AMP [cAMP]-responsive element-binding protein 3-like protein 1 |
| DCTs | dissemination tumor cells |
| Dll4 | notch ligand delta-like ligand 4 |
| ECM | extracellular matrix |
| EGCG | epigallocatechin-3-gallate |
| EGF-SubA | epidermal growth factor-SubA |
| eIF2α | eukaryotic Initiation Factor 2α |
| EMT | epithelial-to-mesenchymal transition |
| eNOS | endothelial nitric oxide synthase |
| EPO | erythropoietin |
| ER | endoplasmic reticulum |
| ERAD | ER-associated degradation |
| ERK | extracellular regulated protein kinases |
| Ero1 | ER oxidoreductin 1 |
| 4μ8C | 8-formyl-7-hydroxy-4-methylcoumarin |
| FGF | fibroblast growth factor |
| FGFBP1 | fibroblast growth factor-binding protein 1 |
| FoxM1 | forkhead box protein M1 |
| HIF | hypoxia-inducible factor |
| 4-HNE | 4-hydroxynonenal |
| IFN-γ | interferon gamma |
| IL-8 | interleukin 8 |
| IRE-1 | Inositol-requiring transmembrane kinase/ endoribonuclease |
| JNK | c-Jun N-terminal kinase |
| MAPK | mitogen-activated protein kinase |
| MEFs | mouse embryonic fibroblasts |
| MHCll | major histocompatibility complex ll |
| MM | human multiple myeloma |
| MMP-2 | matrix metalloproteinases2 |
| MMP-9 | matrix metalloproteinases9 |
| mTOR | Akt-independent mammalian target of rapamycin |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PACMA31 | propynoic acid carbamoyl methyl amide 31 |
| 4-PBA | 4-phenylbutyric acid |
| PDGF | platelet-derived growth factor |
| PERK | protein kinase RNA-activated (PKR)-like ER kinase |
| PFKFB3 | 6-phosphofructo-2-kinase/fructose-2,6- biphosphatase 3 |
| PTN | pleiotrophin |
| Rheb | Ras homolog enriched in brain |
| RIDD | regulating IRE1-dependent decay |
| RIP | regulated intramembrane proteolysis |
| ROS | reactive oxygen species |
| S1P | site-1 protease |
| S2P | site-2 protease |
| Snail | zinc finger protein SNAI1 |
| TDCs | tumor dendritic cells |
| TRAF2 | tumor necrosis factor receptor-associated factor 2 |
| TUD-CA | tauroursodeoxycholic acid |
| uPAR | urokinase-type plasminogen activator receptor |
| UPR | unfolded protein response |
| UPRE | unfolded protein response element |
| VEGF | vascular endothelial growth factor |
References
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, D.S.; Blower, M.D. The endoplasmic reticulum: Structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016, 73, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Namba, T. Regulation of endoplasmic reticulum functions. Aging 2015, 7, 901–902. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Genereux, J.C.; Qu, S.; Zhou, M.; Ryno, L.M.; Wang, S.; Shoulders, M.D.; Kaufman, R.J.; Lasmezas, C.I.; Kelly, J.W.; Wiseman, R.L. Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J. 2015, 34, 4–19. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333. [Google Scholar] [CrossRef]
- Lemus, L.; Goder, V. Regulation of Endoplasmic Reticulum-Associated Protein Degradation (ERAD) by Ubiquitin. Cells 2014, 3, 824–847. [Google Scholar] [CrossRef] [PubMed]
- Vashist, S.; Ng, D.T. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004, 165, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Lee, A.S. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013, 32, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Wang, M.; Law, M.E.; Castellano, R.K.; Law, B.K. The unfolded protein response as a target for anticancer therapeutics. Crit. Rev. Oncol. Hematol. 2018, 127, 66–79. [Google Scholar] [CrossRef]
- Kondo, S.; Saito, A.; Asada, R.; Kanemoto, S.; Imaizumi, K. Physiological unfolded protein response regulated by OASIS family members, transmembrane bZIP transcription factors. IUBMB Life 2011, 63, 233–239. [Google Scholar] [CrossRef]
- Jiang, D.; Niwa, M.; Koong, A.C. Targeting the IRE1alpha-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Zhang, K.Z.; Li, Z.H. Unfolded protein response in cancer: the Physician’s perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef]
- Clarke, H.J.; Chambers, J.E.; Liniker, E.; Marciniak, S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell 2014, 25, 563–573. [Google Scholar] [CrossRef]
- Tirasophon, W.; Lee, K.; Callaghan, B.; Welihinda, A.; Kaufman, R.J. The endoribonuclease activity of mammalian IRE1 autoregulates its mRNA and is required for the unfolded protein response. Genes Dev. 2000, 14, 2725–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.H.; Lee, K.; Vink, E.; Kaufman, R.J. Cytoplasmic IRE1alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J. Biol. Chem. 2006, 281, 18691–18706. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Nishitoh, H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012, 151, 217–219. [Google Scholar] [CrossRef]
- Brush, M.H.; Weiser, D.C.; Shenolikar, S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol. 2003, 23, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, R. ER stress and hepatic lipid metabolism. Front. Genet. 2014, 5, 112. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Lee, B.R.; Chang, S.Y.; Hong, E.H.; Kwon, B.E.; Kim, H.M.; Kim, Y.J.; Lee, J.; Cho, H.J.; Cheon, J.H.; Ko, H.J. Elevated endoplasmic reticulum stress reinforced immunosuppression in the tumor microenvironment via myeloid-derived suppressor cells. Oncotarget 2014, 5, 12331–12345. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; McGrath, E.P.; Mnich, K.; Healy, S.; Chevet, E.; Samali, A. Controlling the unfolded protein response-mediated life and death decisions in cancer. Semin. Cancer Biol. 2015, 33, 57–66. [Google Scholar] [CrossRef]
- Tang, C.H.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C. Inhibition of ER stress-associated IRE-1/XBP-1 pathway reduces leukemic cell survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranganathan, A.C.; Adam, A.P.; Zhang, L.; Aguirre-Ghiso, J.A. Tumor cell dormancy induced by p38SAPK and ER-stress signaling: An adaptive advantage for metastatic cells? Cancer Biol. Ther. 2006, 5, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Chitnis, N.S.; Pytel, D.; Bobrovnikova-Marjon, E.; Pant, D.; Zheng, H.; Maas, N.L.; Frederick, B.; Kushner, J.A.; Chodosh, L.A.; Koumenis, C.; et al. miR-211 is a prosurvival microRNA that regulates chop expression in a PERK-dependent manner. Mol. Cell 2012, 48, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Pfaffenbach, K.T.; Lee, A.S. The critical role of GRP78 in physiologic and pathologic stress. Curr. Opin. Cell Biol. 2011, 23, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Zhou, L.; Xia, M.H.; Xu, Y.; Xiang, X.Y.; Sun, L.K. Bcl-2 family proteins are involved in the signal crosstalk between endoplasmic reticulum stress and mitochondrial dysfunction in tumor chemotherapy resistance. Biomed Res. Int. 2014, 2014, 234370. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Vanacker, H.; Vetters, J.; Moudombi, L.; Caux, C.; Janssens, S.; Michallet, M.C. Emerging Role of the Unfolded Protein Response in Tumor Immunosurveillance. Trends Cancer 2017, 3, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera-Ramirez, L.; Hunter, K.W. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Research 2017, 6, 2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paez, D.; Labonte, M.J.; Bohanes, P.; Zhang, W.; Benhanim, L.; Ning, Y.; Wakatsuki, T.; Loupakis, F.; Lenz, H.J. Cancer dormancy: A model of early dissemination and late cancer recurrence. Clin. Cancer Res. 2012, 18, 645–653. [Google Scholar] [CrossRef]
- Gao, X.L.; Zhang, M.; Tang, Y.L.; Liang, X.H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. Oncotargets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2(+) mammary cancer. Nature 2016. [Google Scholar] [CrossRef]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef]
- Sosa, M.S.; Bragado, P.; Debnath, J.; Aguirre-Ghiso, J.A. Regulation of tumor cell dormancy by tissue microenvironments and autophagy. Adv. Exp. Med. Biol. 2013, 734, 73–89. [Google Scholar]
- Ranganathan, A.C.; Ojha, S.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Dual function of pancreatic endoplasmic reticulum kinase in tumor cell growth arrest and survival. Cancer Res. 2008, 68, 3260–3268. [Google Scholar] [CrossRef]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase-like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Lu, Z.H.; Shvartsman, M.B.; Lee, A.Y.; Shao, J.M.; Murray, M.M.; Kladney, R.D.; Fan, D.; Krajewski, S.; Chiang, G.G.; Mills, G.B.; et al. Mammalian target of rapamycin activator RHEB is frequently overexpressed in human carcinomas and is critical and sufficient for skin epithelial carcinogenesis. Cancer Res. 2010, 70, 3287–3298. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Semmling, V.; Franken, L.; Wagner, H.; Kurts, C. Chemokines: A new dendritic cell signal for T cell activation. Front. Immunol. 2011, 2, 31. [Google Scholar] [CrossRef]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Annu. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Molecular Pathways: Immunosuppressive Roles of IRE1alpha-XBP1 Signaling in Dendritic Cells of the Tumor Microenvironment. Clin. Cancer Res. 2016, 22, 2121–2126. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Hetz, C. A novel ER stress-independent function of the UPR in angiogenesis. Mol. Cell 2014, 54, 542–544. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.R.; Frudd, K.; Awad, W.; Hendershot, L.M. Endoplasmic reticulum (ER) stress and hypoxia response pathways interact to potentiate hypoxia-inducible factor 1 (HIF-1) transcriptional activity on targets like vascular endothelial growth factor (VEGF). J. Biol. Chem. 2014, 289, 3352–3364. [Google Scholar] [CrossRef] [PubMed]
- Hanania, R.; Sun, H.S.; Xu, K.; Pustylnik, S.; Jeganathan, S.; Harrison, R.E. Classically activated macrophages use stable microtubules for matrix metalloproteinase-9 (MMP-9) secretion. J. Biol. Chem. 2012, 287, 8468–8483. [Google Scholar] [CrossRef]
- Gee, E.; Milkiewicz, M.; Haas, T.L. p38 MAPK activity is stimulated by vascular endothelial growth factor receptor 2 activation and is essential for shear stress-induced angiogenesis. J. Cell. Physiol. 2010, 222, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 2010, 5, e9575. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Yamanaka, S.; Kohno, K. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. Proc. Natl. Acad. Sci. USA 2009, 106, 16657–16662. [Google Scholar] [CrossRef] [Green Version]
- Oskolkova, O.V.; Afonyushkin, T.; Leitner, A.; von Schlieffen, E.; Gargalovic, P.S.; Lusis, A.J.; Binder, B.R.; Bochkov, V.N. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: Critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood 2008, 112, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Dubey, S.; Varney, M.L.; Dave, B.J.; Singh, R.K. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J. Immunol. 2003, 170, 3369–3376. [Google Scholar] [CrossRef]
- Zeng, L.; Xiao, Q.; Chen, M.; Margariti, A.; Martin, D.; Ivetic, A.; Xu, H.; Mason, J.; Wang, W.; Cockerill, G.; et al. Vascular endothelial cell growth-activated XBP1 splicing in endothelial cells is crucial for angiogenesis. Circulation 2013, 127, 1712–1722. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Fernandez-Checa, J.C.; Kaplowitz, N. JNK interaction with Sab mediates ER stress induced inhibition of mitochondrial respiration and cell death. Cell Death Dis. 2014, 5, e989. [Google Scholar] [CrossRef]
- Loinard, C.; Zouggari, Y.; Rueda, P.; Ramkhelawon, B.; Cochain, C.; Vilar, J.; Recalde, A.; Richart, A.; Charue, D.; Duriez, M.; et al. C/EBP homologous protein-10 (CHOP-10) limits postnatal neovascularization through control of endothelial nitric oxide synthase gene expression. Circulation 2012, 125, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Mellor, P.; Deibert, L.; Calvert, B.; Bonham, K.; Carlsen, S.A.; Anderson, D.H. CREB3L1 is a metastasis suppressor that represses expression of genes regulating metastasis, invasion, and angiogenesis. Mol. Cell. Biol. 2013, 33, 4985–4995. [Google Scholar] [CrossRef]
- Binet, F.; Sapieha, P. ER Stress and Angiogenesis. Cell Metab. 2015, 22, 560–575. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Feng, Y.X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed]
- Denard, B.; Lee, C.; Ye, J. Doxorubicin blocks proliferation of cancer cells through proteolytic activation of CREB3L1. eLife 2012, 1, e00090. [Google Scholar] [CrossRef]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534. [Google Scholar] [CrossRef]
- Shi, S.; Tan, P.; Yan, B.; Gao, R.; Zhao, J.; Wang, J.; Guo, J.; Li, N.; Ma, Z. ER stress and autophagy are involved in the apoptosis induced by cisplatin in human lung cancer cells. Oncol. Rep. 2016, 35, 2606–2614. [Google Scholar] [CrossRef]
- Chong, D.C.; Paton, J.C.; Thorpe, C.M.; Paton, A.W. Clathrin-dependent trafficking of subtilase cytotoxin, a novel AB5 toxin that targets the endoplasmic reticulum chaperone BiP. Cell. Microbiol. 2008, 10, 795–806. [Google Scholar] [CrossRef]
- Virrey, J.J.; Dong, D.; Stiles, C.; Patterson, J.B.; Pen, L.; Ni, M.; Schonthal, A.H.; Chen, T.C.; Hofman, F.M.; Lee, A.S. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res. 2008, 6, 1268–1275. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Gao, W.; Zhou, Y.; Wey, S.; Mitra, S.K.; Krasnoperov, V.; Dong, D.; Liu, S.; Li, D.; et al. Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin. Cancer Res. 2013, 19, 6802–6811. [Google Scholar] [CrossRef]
- Rosenes, Z.; Mulhern, T.D.; Hatters, D.M.; Ilag, L.L.; Power, B.E.; Hosking, C.; Hensel, F.; Howlett, G.J.; Mok, Y.F. The anti-cancer IgM monoclonal antibody PAT-SM6 binds with high avidity to the unfolded protein response regulator GRP78. PLoS ONE 2012, 7, e44927. [Google Scholar] [CrossRef] [PubMed]
- Rasche, L.; Duell, J.; Morgner, C.; Chatterjee, M.; Hensel, F.; Rosenwald, A.; Einsele, H.; Topp, M.S.; Brandlein, S. The natural human IgM antibody PAT-SM6 induces apoptosis in primary human multiple myeloma cells by targeting heat shock protein GRP78. PLoS ONE 2013, 8, e63414. [Google Scholar] [CrossRef]
- Glaser, K.B. HDAC inhibitors: Clinical update and mechanism-based potential. Biochem. Pharmacol. 2007, 74, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. eLife 2016, 5, e11878. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.M.; Walter, P. Ceapins inhibit ATF6alpha signaling by selectively preventing transport of ATF6alpha to the Golgi apparatus during ER stress. eLife 2016, 5, e11880. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Haze, K.; Nadanaka, S.; Yoshida, H.; Seidah, N.G.; Hirano, Y.; Sato, R.; Negishi, M.; Mori, K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem. 2003, 278, 31024–31032. [Google Scholar] [CrossRef]
- Higa, A.; Taouji, S.; Lhomond, S.; Jensen, D.; Fernandez-Zapico, M.E.; Simpson, J.C.; Pasquet, J.M.; Schekman, R.; Chevet, E. Endoplasmic reticulum stress-activated transcription factor ATF6alpha requires the disulfide isomerase PDIA5 to modulate chemoresistance. Mol. Cell. Biol. 2014, 34, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Vincenz, L.; Jager, R.; O’Dwyer, M.; Samali, A. Endoplasmic reticulum stress and the unfolded protein response: Targeting the Achilles heel of multiple myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Rivera, D.; Delvaeye, T.; Roelandt, R.; Nerinckx, W.; Augustyns, K.; Vandenabeele, P.; Bertrand, M.J.M. When PERK inhibitors turn out to be new potent RIPK1 inhibitors: Critical issues on the specificity and use of GSK2606414 and GSK2656157. Cell Death Differ. 2017, 24, 1100–1110. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Gaedicke, S.; Firat-Geier, E.; Constantiniu, O.; Lucchiari-Hartz, M.; Freudenberg, M.; Galanos, C.; Niedermann, G. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: Induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002, 62, 6901–6908. [Google Scholar] [PubMed]
- Wang, G.; Yang, Z.Q.; Zhang, K. Endoplasmic reticulum stress response in cancer: Molecular mechanism and therapeutic potential. Am. J. Transl. Res. 2010, 2, 65–74. [Google Scholar]
- Adam, C.; Baeurle, A.; Brodsky, J.L.; Wipf, P.; Schrama, D.; Becker, J.C.; Houben, R. The HSP70 modulator MAL3-101 inhibits Merkel cell carcinoma. PLoS ONE 2014, 9, e92041. [Google Scholar]
- Bachleitner-Hofmann, T.; Sun, M.Y.; Chen, C.T.; Liska, D.; Zeng, Z.; Viale, A.; Olshen, A.B.; Mittlboeck, M.; Christensen, J.G.; Rosen, N.; et al. Antitumor activity of SNX-2112, a synthetic heat shock protein-90 inhibitor, in MET-amplified tumor cells with or without resistance to selective MET Inhibition. Clin. Cancer Res. 2011, 17, 122–133. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Ye, Q.; Scott, A.; Silinski, M.; Huang, K.; Fadden, P.; Partdrige, J.; Hall, S.; Steed, P.; et al. SNX2112, a synthetic heat shock protein 90 inhibitor, has potent antitumor activity against HER kinase-dependent cancers. Clin. Cancer Res. 2008, 14, 240–248. [Google Scholar] [CrossRef]
- Pyrko, P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M.; Krivoshein, A.V.; Hamby, C.V.; Pizzonia, J.; Gilbert, K.S.; Ray, Y.S.; Brand, H.; Paton, A.W.; Paton, J.C.; Backer, M.V. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia 2009, 11, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Martinotti, S.; Ranzato, E.; Burlando, B. (−)- Epigallocatechin-3-gallate induces GRP78 accumulation in the ER and shifts mesothelioma constitutive UPR into proapoptotic ER stress. J. Cell. Physiol. 2018, 233, 7082–7090. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhu, C.; Song, D.; Xia, R.; Yu, W.; Dang, Y.; Fei, Y.; Yu, L.; Wu, J. Epigallocatechin-3-gallate enhances ER stress-induced cancer cell apoptosis by directly targeting PARP16 activity. Cell Death Discov. 2017, 3, 17034. [Google Scholar] [CrossRef]
- Ermakova, S.P.; Kang, B.S.; Choi, B.Y.; Choi, H.S.; Schuster, T.F.; Ma, W.Y.; Bode, A.M.; Dong, Z. (−)-Epigallocatechin gallate overcomes resistance to etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006, 66, 9260–9269. [Google Scholar] [CrossRef]
- Min, K.J.; Kwon, T.K. Anticancer effects and molecular mechanisms of epigallocatechin-3-gallate. Integr. Med. Res. 2014, 3, 16–24. [Google Scholar] [CrossRef]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Ali, M.M.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.; Estrada, A.; Kim, P.; Wang, D.; Wei, Y.; Gentile, C.; Pagliassotti, M. Regulation of IRE1alpha by the small molecule inhibitor 4mu8c in hepatoma cells. Endoplasmic Reticulum Stress Dis. 2017, 4, 1–10. [Google Scholar] [CrossRef]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2 alpha. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Romeril, S.P.; Shu, A.; Ralph, J.; Medina, J.R.; Feng, Y.; Li, W.H.; Grant, S.W.; Heerding, D.A.; Minthorn, E.; et al. Discovery of GSK2656157: An Optimized PERK Inhibitor Selected for Preclinical Development. ACS Med. Chem. Lett. 2013, 4, 964–968. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Gao, M.; Wang, J.; Kong, Q.; Hua, H.; Luo, T.; Jiang, Y. Inhibition of eIF2alpha dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis. 2014, 5, e1060. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Kim, J.H.; Shin, J.I.; Jeong, M.; Cho, J.; Lee, K. Salubrinal-Mediated Upregulation of eIF2alpha Phosphorylation Increases Doxorubicin Sensitivity in MCF-7/ADR Cells. Mol. Cells 2016, 39, 129–135. [Google Scholar]
- Friedman, J.A.; Wise, S.C.; Hu, M.; Gouveia, C.; Vander Broek, R.; Freudlsperger, C.; Kannabiran, V.R.; Arun, P.; Mitchell, J.B.; Chen, Z.; et al. HSP90 Inhibitor SNX5422/2112 Targets the Dysregulated Signal and Transcription Factor Network and Malignant Phenotype of Head and Neck Squamous Cell Carcinoma. Transl. Oncol. 2013, 6, 429–441. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stage | Target | Mechanism | Small Molecule | Outcome | Reference | |
|---|---|---|---|---|---|---|
| Initiation | Grp78 (Bip) | Unknown | 4-Phenyl-butyric acid (4-PBA); Tauroursodeoxycholic acid (TUDC) | Blocking ER stress to induce cytotoxicity and apoptosis | [84,85,86] | |
| Specifically cleaving Grp78 at a di-leucine motif | EGF-SubA | Leading to high cytotoxicity and reducing chemo-resistance | [11,87] | |||
| Binding to the ATP-binding site of Grp78 and modulating its ATPase activity | Epigallocatechin-3-gallate (EGCG) | Enhancing ER stress-induced cancer cell apoptosis | [10] | |||
| Inhibiting the ATPase activity of Grp78 | CPT-11, etoposide, and temozolomide | Increasing sensitivity of cancer cells to bortezomib | [88] | |||
| Triggering Grp78 endocytosis | Mouse monoclonal antibody (mAb159) | Inhibiting endothelial cells and angiogenesis | [89] | |||
| Recognizing tumor cells expressing Grp78 and inducing complement dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC) | PAT-SM6 (monoclonal IgM) | Inducing MM cell death | [90,91] | |||
| Blocking the histone deacetylase and bringing about acetylation of Grp78 | Vorinostat (HDAC Inhibitors) | Intruding the function of Grp78, which contributes to the accumulation of misfolded protein and cell death | [92] | |||
| Sensor | ATF6 | Directly target ATF6 | Selectively blocking ATF6 and trapping it in the ER | Ceapins | Sensitizing cancer cells to ER stress | [93,94] |
| Indirectly target ATF6, mainly target associated enzymes | Hindering the proteases S1P and S2P in the Golgi body | 4-(2-Aminoethyl) benzene- sulfonyl fluoride (AEBSP) | Blocking nuclear localization and inhibiting ATF6 downstream signaling | [95] | ||
| Unknown | Propynoic acid carbamoyl methyl amide 31 (PACMA31) | Block ATF6 downstream signaling | [96] | |||
| Inhibit the disulfide bond formation of ATF6 | 16 F16 | Reducing the chemo-resistance and promoting sensitivity to Imatinib | [96] | |||
| Sensor | IRE-1-XBP1 | Interacting with the catalytic core of the RNase domain of IRE-1α | 8-Formyl-7-hydroxy-4-methyl coumarin (4μ8c) | Inhibiting the endoribonuclease (RNase) activity of IRE1 | [11,13,97] | |
| MKC-3946 | Inhibiting RNase activity of IRE-1 and increase expression of CHOP | [11,13,97] | ||||
| Binding with the ATP binding site within the IRE-1 kinase domain | APY29, Sunitinib | Inhibiting IRE-1 phosphorylation and indirectly suppressing its RNase activity | [11,13] | |||
| Stabilizing the inactive conformation of the ATP-binding site within the IRE-1 kinase domain | Quercetin | |||||
| PERK | Competing with the ATP-binding site within PERK kinase | GSK2656157 | Inhibiting PERK autophosphorylation and phosphorylation of eIF2α | [98] | ||
| Downstream | eIF2α | Interrupting the activity of GADD34/PP1c complex and protecting eIF2α from dephosphorylation | Salubrinal; Guanabenz | Stimulating eIF2α phosphorylation, inducing expression of CHOP and trigger apoptosis | [99,100] | |
| ERAD | ERAD | Blocking the 26S proteasome and intrude proteolysis | Bortezomib | Inhibiting ERAD and retarding the proliferation of cancer cells | [101,102] | |
| Ritonavir | Crippling the ERAD system, and causing misfolded protein overloading | [102,103] | ||||
| Interacting with p97 ATPase and block ERAD | Eeyarestatin I | Triggering NOXA and inducing cancer cell apoptosis | [104] | |||
| Chaperone | HSP70 | Interfering with the ATPase activity of HSP70 proteins | MAL3-101 | Blocking the function of HSP70, which leads to the accumulation of misfolded protein and apoptosis | [105] | |
| HSP90 | Competitively interacting with the N-terminal ATP-binding site of HSP90 | Retaspimycin (IPI-504) | Leading to the instability of oncogenic kinases and bringing about cell cycle arrest or apoptosis | [106,107] | ||
| SNX-2112 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, S.-K.; Chiu, C.-C.; Dahms, H.-U.; Chou, C.-K.; Cheng, C.-M.; Chang, W.-T.; Cheng, K.-C.; Wang, H.-M.D.; Lin, I.-L. Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2518. https://doi.org/10.3390/ijms20102518
Hsu S-K, Chiu C-C, Dahms H-U, Chou C-K, Cheng C-M, Chang W-T, Cheng K-C, Wang H-MD, Lin I-L. Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells. International Journal of Molecular Sciences. 2019; 20(10):2518. https://doi.org/10.3390/ijms20102518
Chicago/Turabian StyleHsu, Sheng-Kai, Chien-Chih Chiu, Hans-Uwe Dahms, Chon-Kit Chou, Chih-Mei Cheng, Wen-Tsan Chang, Kai-Chun Cheng, Hui-Min David Wang, and I-Ling Lin. 2019. "Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells" International Journal of Molecular Sciences 20, no. 10: 2518. https://doi.org/10.3390/ijms20102518
APA StyleHsu, S.-K., Chiu, C.-C., Dahms, H.-U., Chou, C.-K., Cheng, C.-M., Chang, W.-T., Cheng, K.-C., Wang, H.-M. D., & Lin, I.-L. (2019). Unfolded Protein Response (UPR) in Survival, Dormancy, Immunosuppression, Metastasis, and Treatments of Cancer Cells. International Journal of Molecular Sciences, 20(10), 2518. https://doi.org/10.3390/ijms20102518