The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Ameliorates High-fat Induced Cognitive Decline in Tauopathy Model Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
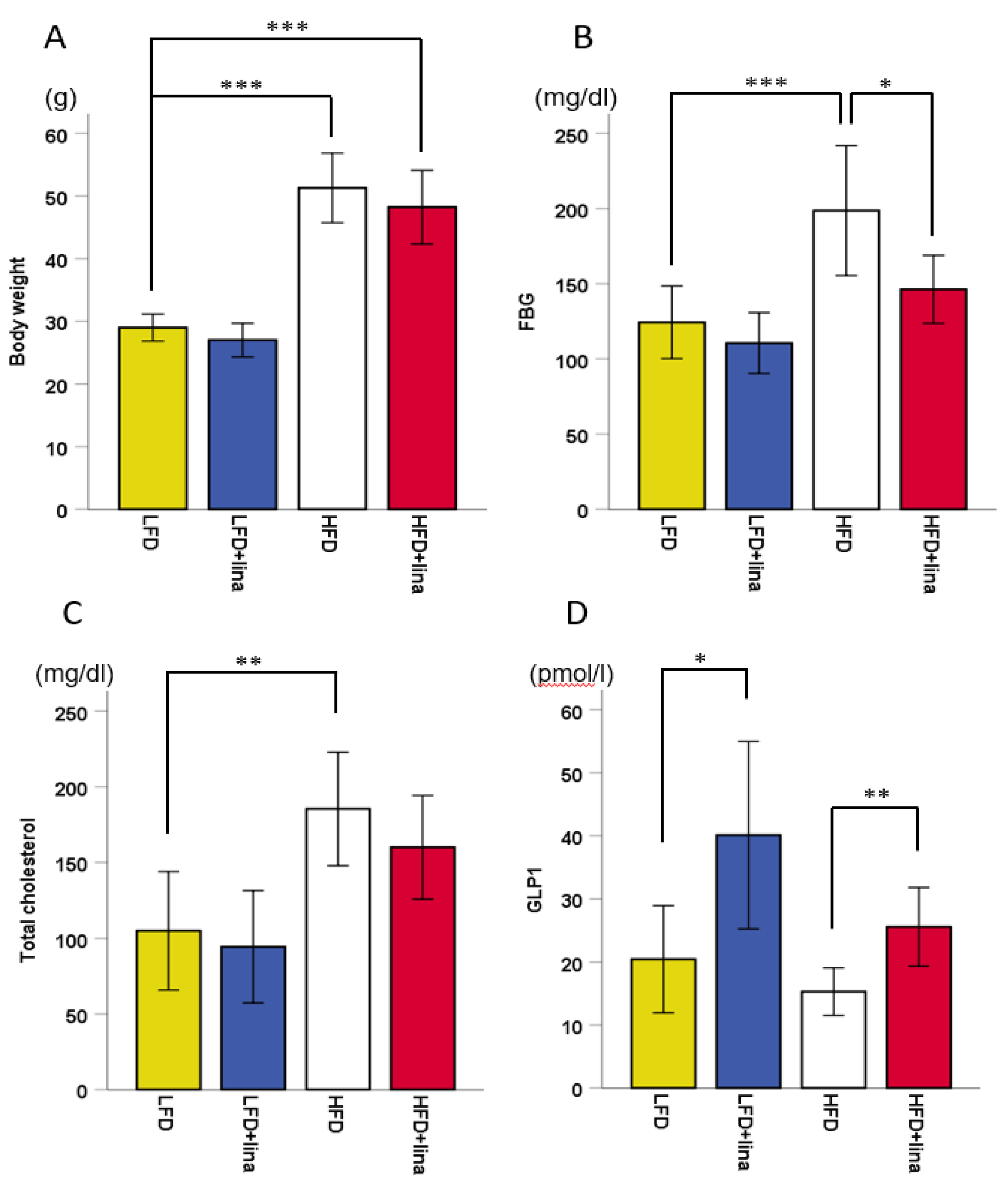
2.1. Hyperglycemia by HFD was Ameliorated by Linagliptin Treatment
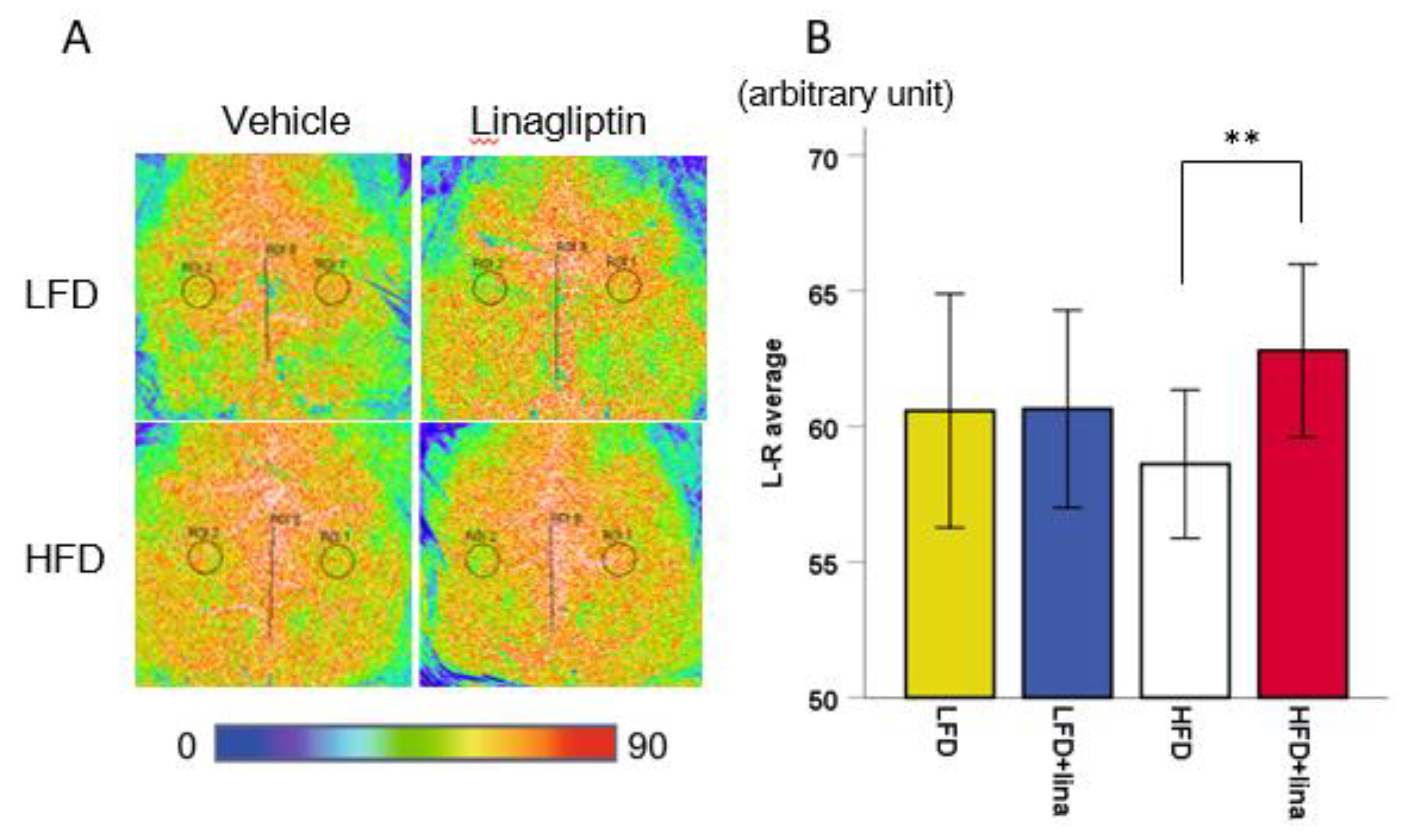
2.2. Restoration of CBF in Linagliptin-Treated PS 19 Mice
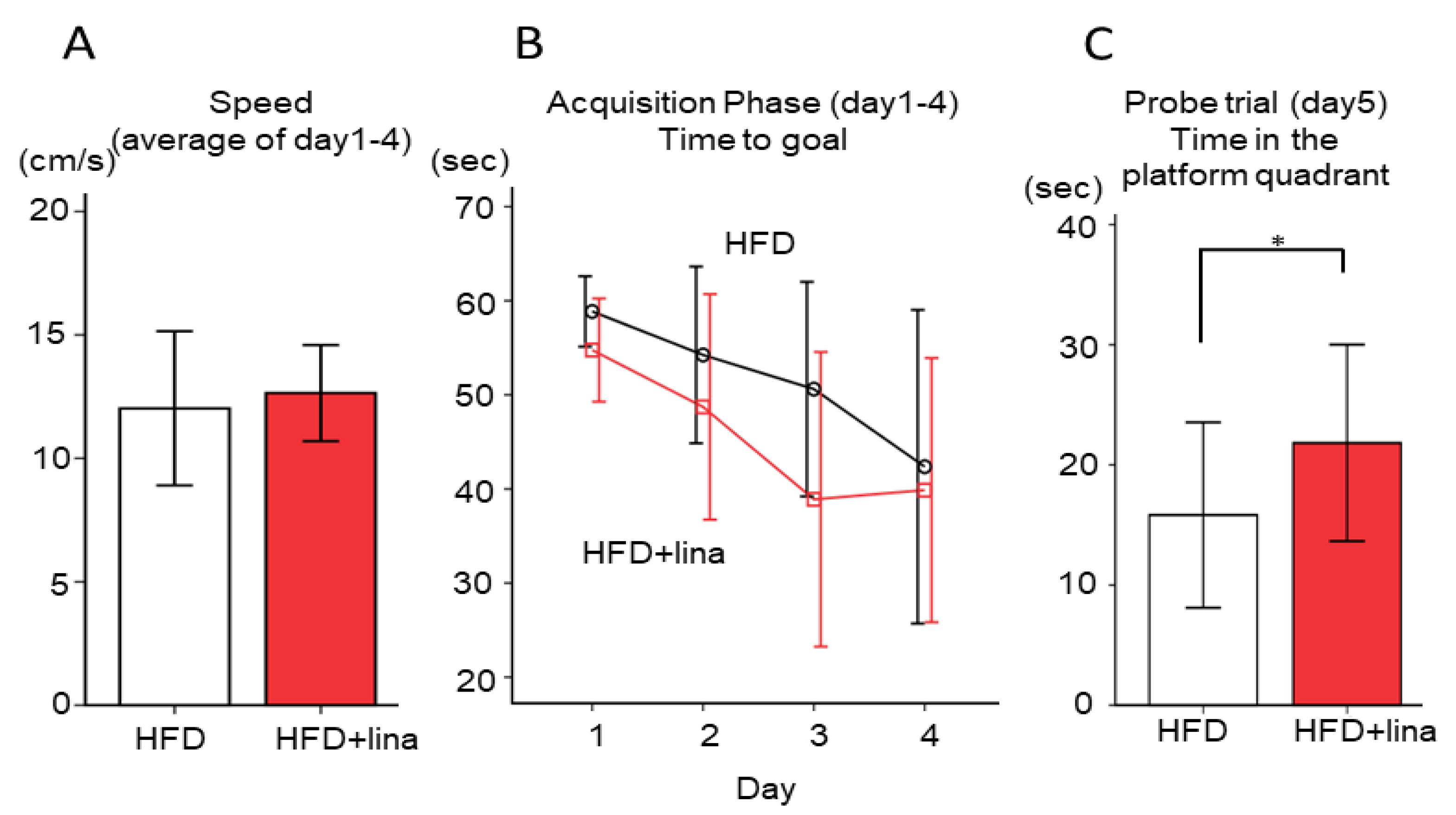
2.3. Normalization of Spatial Reference Memory Impairment in HFD-fed PS19 Mice .
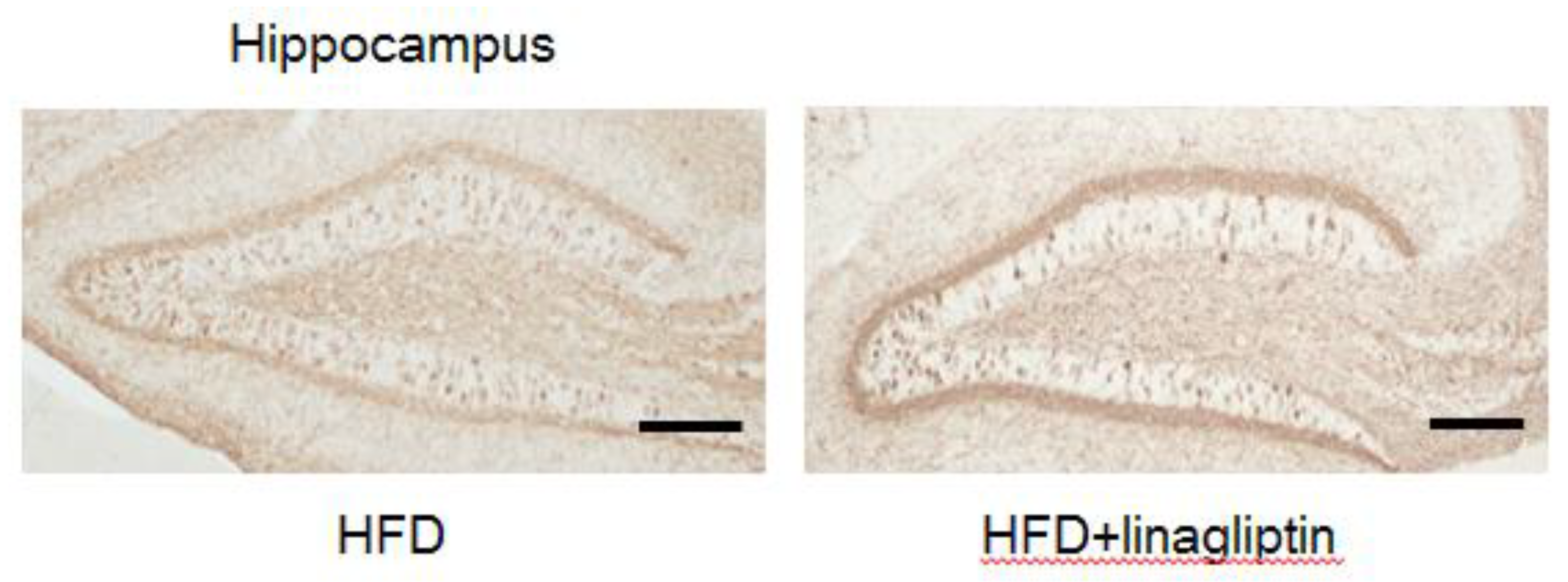
2.4. Immunohistochemistry
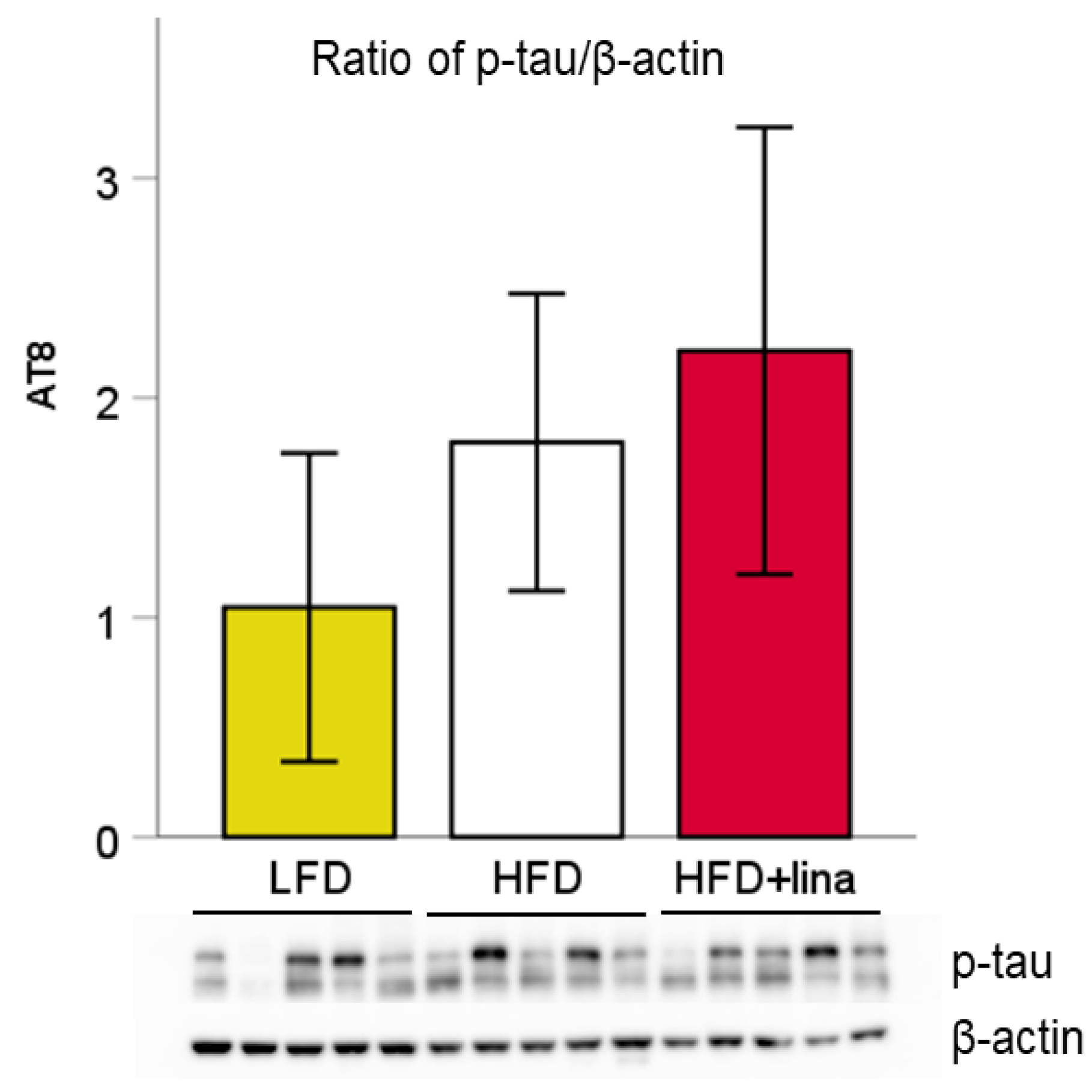
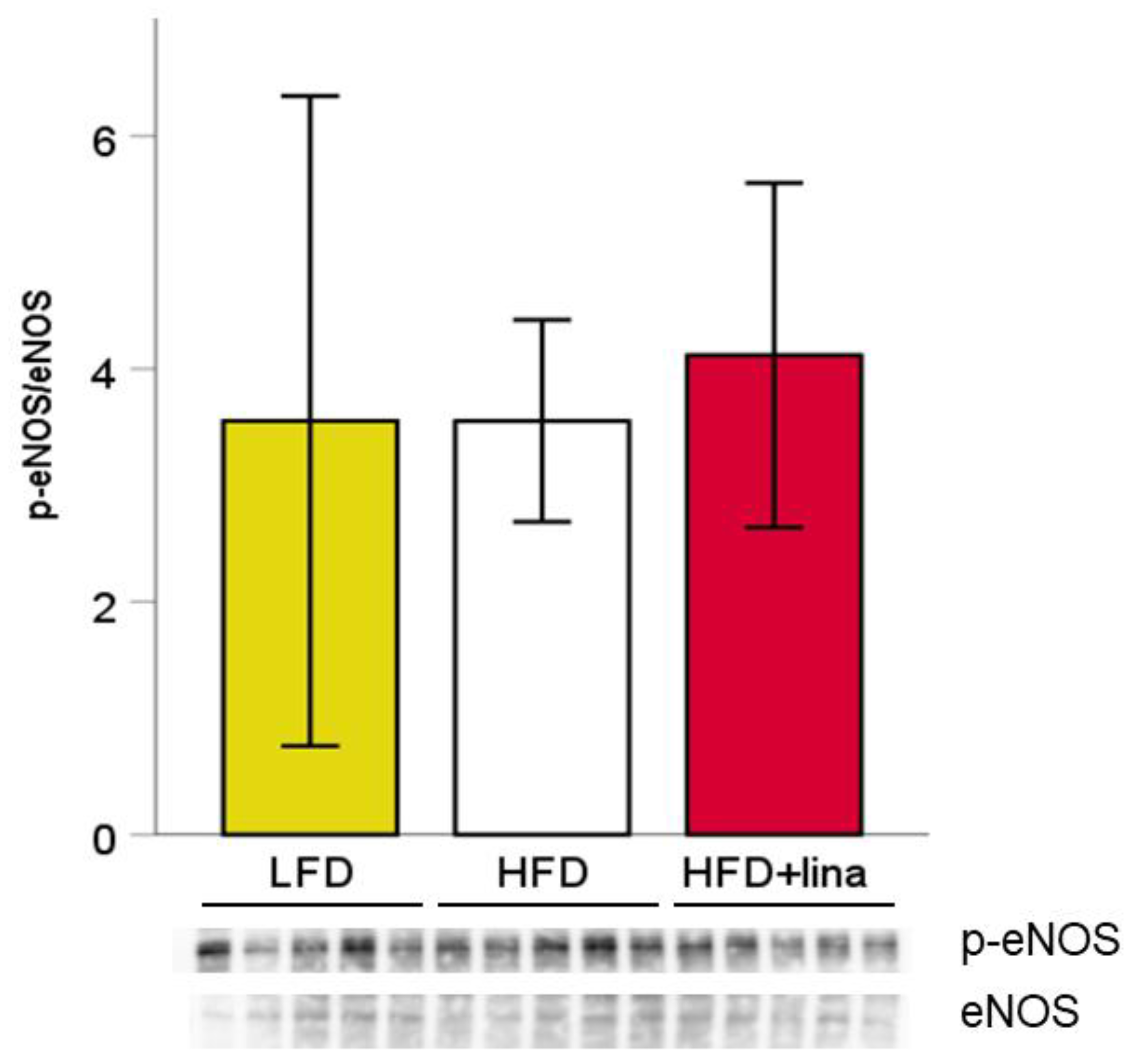
2.5. Western Blotting
3. Discussion
4. Materials and Methods
4.1. Animals
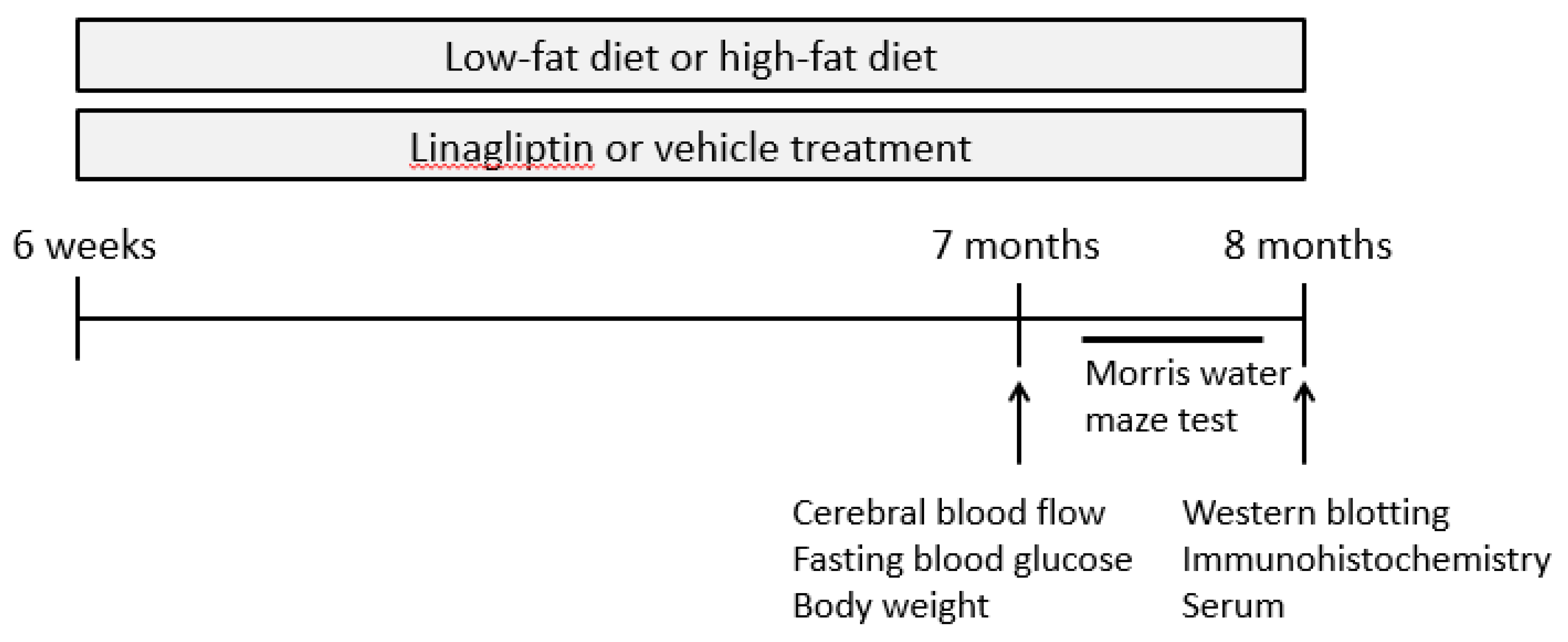
4.2. Study Design
4.3. Blood Testing
4.4. Measurement of Cerebral Blood Flow
4.5. Behavioral Analysis: Morris Water Maze Test
4.6. Western Blotting
4.7. Immunohistochemistry
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| T2DM | Type 2 diabetes mellitus |
| DPP-4 | Dipeptidyl peptidase-4 |
| HFD | High-fat diet |
| LFD | Low-fat diet |
| AD | Alzheimer’s disease |
| Aβ | β-amyloid |
| NFT | neurofibrillary tangle |
| GLP-1 | glucagon-like peptide-1 |
| FTDP-17 | frontotemporal dementia and parkinsonism linked to chromosome 17 |
| CBF | cerebral blood flow |
| MAPT | microtubule-associated protein tau |
| Prnp | prion protein promoter |
| PCR | polymerase chain reaction |
| MWM | Morris water maze test |
| NO | nitric oxide |
| EDHF | endothelium-derived hyperpolarizing factor |
| eNOS | endothelial nitric oxide synthase |
| PBS | phosphate buttered saline |
| RIPA | radio-immunoprecipitation assay |
| PVDF | polyvinylidene difluoride |
| DAB | diaminobenzidine |
References
- Kumar, A.; Singh, A.; Ekavali. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992, 42, 631–639. [Google Scholar] [CrossRef]
- Koekkoek, P.S.; Kappelle, L.J.; van den Berg, E.; Rutten, G.E.; Biessels, G.J. Cognitive function in patients with diabetes mellitus: guidance for daily care. Lancet Neurol. 2015, 14, 329–340. [Google Scholar] [CrossRef]
- Alam, F.; Islam, M.A.; Sasongko, T.H.; Gan, S.H. Type 2 Diabetes Mellitus and Alzheimer’s Disease: Bridging the Pathophysiology and Management. Curr. Pharm. Des. 2016, 22, 4430–4442. [Google Scholar] [CrossRef]
- Bedse, G.; Di Domenico, F.; Serviddio, G.; Cassano, T. Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci. 2015, 9, 204. [Google Scholar] [CrossRef]
- Gupta, A.; Bisht, B.; Dey, C.S. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology 2011, 60, 910–920. [Google Scholar] [CrossRef]
- El Khoury, N.B.; Gratuze, M.; Papon, M.A.; Bretteville, A.; Planel, E. Insulin dysfunction and Tau pathology. Front. Cell. Neurosci. 2014, 8, 22. [Google Scholar] [CrossRef]
- Yue, J.T.; Lam, T.K. Lipid sensing and insulin resistance in the brain. Cell Metab. 2012, 15, 646–655. [Google Scholar] [CrossRef]
- Levin-Allerhand, J.A.; Lominska, C.E.; Smith, J.D. Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J. Nutr. Health Aging 2002, 6, 315–319. [Google Scholar]
- Leboucher, A.; Laurent, C.; Fernandez-Gomez, F.J.; Burnouf, S.; Troquier, L.; Eddarkaoui, S.; Demeyer, D.; Caillierez, R.; Zommer, N.; Vallez, E.; et al. Detrimental effects of diet-induced obesity on τ pathology are independent of insulin resistance in τ transgenic mice. Diabetes 2013, 62, 1681–1688. [Google Scholar] [CrossRef]
- Lester-Coll, N.; Rivera, E.J.; Soscia, S.J.; Doiron, K.; Wands, J.R.; de la Monte, S.M. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 13–33. [Google Scholar] [CrossRef]
- de la Monte, S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef]
- Yarchoan, M.; Arnold, S.E. Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease. Diabetes 2014, 63, 2253–2261. [Google Scholar] [CrossRef]
- Ahrén, B.; Schmitz, O. GLP-1 receptor agonists and DPP-4 inhibitors in the treatment of type 2 diabetes. Horm. Metab. Res. 2004, 36, 867–876. [Google Scholar] [CrossRef]
- Sebastião, I.; Candeias, E.; Santos, M.S.; de Oliveira, C.R.; Moreira, P.I.; Duarte, A.I. Insulin as a Bridge between Type 2 Diabetes and Alzheimer Disease - How Anti-Diabetics Could be a Solution for Dementia. Front. Endocrinol. (Lausanne). 2014, 5, 110. [Google Scholar] [CrossRef]
- Hamilton, A.; Patterson, S.; Porter, D.; Gault, V.A.; Holscher, C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J. Neurosci. Res. 2011, 89, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Harkavyi, A.; Whitton, P.S. Glucagon-like peptide 1 receptor stimulation as a means of neuroprotection. Br. J. Pharmacol. 2010, 159, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holscher, C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer’s disease. Recent Pat. CNS Drug Discov. 2010, 5, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y. The nature, significance, and glucagon-like peptide-1 analog treatment of brain insulin resistance in Alzheimer’s disease. Alzheimers Dement. 2014, 10, S12–S25. [Google Scholar] [CrossRef] [PubMed]
- Long-Smith, C.M.; Manning, S.; McClean, P.L.; Coakley, M.F.; O’Halloran, D.J.; Holscher, C.; O’Neill, C. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-β plaque and glial pathology in a mouse model of Alzheimer’s disease. Neuromolecular Med. 2013, 15, 102–114. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Hölscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. 2014, 76 Pt A, 57–67. [Google Scholar] [CrossRef]
- Kosaraju, J.; Holsinger, R.M.D.; Guo, L.; Tam, K.Y. Linagliptin, a Dipeptidyl Peptidase-4 Inhibitor, Mitigates Cognitive Deficits and Pathology in the 3xTg-AD Mouse Model of Alzheime’s Disease. Mol. Neurobiol. 2017, 54, 6074–6084. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Barkholt, P.; Fabricius, K.; Jelsing, J.; Terwel, D.; Pyke, C.; Knudsen, L.B.; Vrang, N. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res. 2016, 1634, 158–170. [Google Scholar] [CrossRef]
- Kim, D.H.; Huh, J.W.; Jang, M.; Suh, J.H.; Kim, T.W.; Park, J.S.; Yoon, S.Y. Sitagliptin increases tau phosphorylation in the hippocampus of rats with type 2 diabetes and in primary neuron cultures. Neurobiol. Dis. 2012, 46, 52–58. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Hayashi, K.; Takemoto, Y.; Cheng, C.; Takane, K.; Lin, B.; Komohara, Y.; Kim-Mitsuyama, S. DPP-4 inhibition with linagliptin ameliorates the progression of premature aging in klotho-/- mice. Cardiovasc. Diabetol. 2017, 16, 154. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Kröller-Schön, S.; Knorr, M.; Hausding, M.; Oelze, M.; Schuff, A.; Schell, R.; Sudowe, S.; Scholz, A.; Daub, S.; Karbach, S.; et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc. Res. 2012, 96, 140–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardigan, T.; Abdul, Y.; Ergul, A. Linagliptin reduces effects of ET-1 and TLR2-mediated cerebrovascular hyperreactivity in diabetes. Life Sci. 2016, 159, 90–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gault, V.A.; Lennox, R.; Flatt, P.R. Sitagliptin, a dipeptidyl peptidase-4 inhibitor, improves recognition memory, oxidative stress and hippocampal neurogenesis and upregulates key genes involved in cognitive decline. Diabetes Obes. Metab. 2015, 17, 403–413. [Google Scholar] [CrossRef]
- Ma, M.; Hasegawa, Y.; Koibuchi, N.; Toyama, K.; Uekawa, K.; Nakagawa, T.; Lin, B.; Kim-Mitsuyama, S. DPP-4 inhibition with linagliptin ameliorates cognitive impairment and brain atrophy induced by transient cerebral ischemia in type 2 diabetic mice. Cardiovasc. Diabetol. 2015, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Enmi, J.; Kitamura, A.; Yamamoto, Y.; Saito, S.; Takahashi, Y.; Iguchi, S.; Tsuji, M.; Yamahara, K.; Nagatsuka, K.; et al. A novel mouse model of subcortical infarcts with dementia. J. Neurosci. 2015, 35, 3915–3928. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakaoku, Y.; Saito, S.; Yamamoto, Y.; Maki, T.; Takahashi, R.; Ihara, M. The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Ameliorates High-fat Induced Cognitive Decline in Tauopathy Model Mice. Int. J. Mol. Sci. 2019, 20, 2539. https://doi.org/10.3390/ijms20102539
Nakaoku Y, Saito S, Yamamoto Y, Maki T, Takahashi R, Ihara M. The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Ameliorates High-fat Induced Cognitive Decline in Tauopathy Model Mice. International Journal of Molecular Sciences. 2019; 20(10):2539. https://doi.org/10.3390/ijms20102539
Chicago/Turabian StyleNakaoku, Yuriko, Satoshi Saito, Yumi Yamamoto, Takakuni Maki, Ryosuke Takahashi, and Masafumi Ihara. 2019. "The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Ameliorates High-fat Induced Cognitive Decline in Tauopathy Model Mice" International Journal of Molecular Sciences 20, no. 10: 2539. https://doi.org/10.3390/ijms20102539
APA StyleNakaoku, Y., Saito, S., Yamamoto, Y., Maki, T., Takahashi, R., & Ihara, M. (2019). The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Ameliorates High-fat Induced Cognitive Decline in Tauopathy Model Mice. International Journal of Molecular Sciences, 20(10), 2539. https://doi.org/10.3390/ijms20102539