Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model

 , ,
, , 

Abstract
:1. Introduction
2. Results
2.1. Altered Liquoral Levels of H2S in Male and Female ALS Patients
2.2. Effects of AOA Treatment on H2S Production in Primary SOD1G93A Spinal Cord Culture
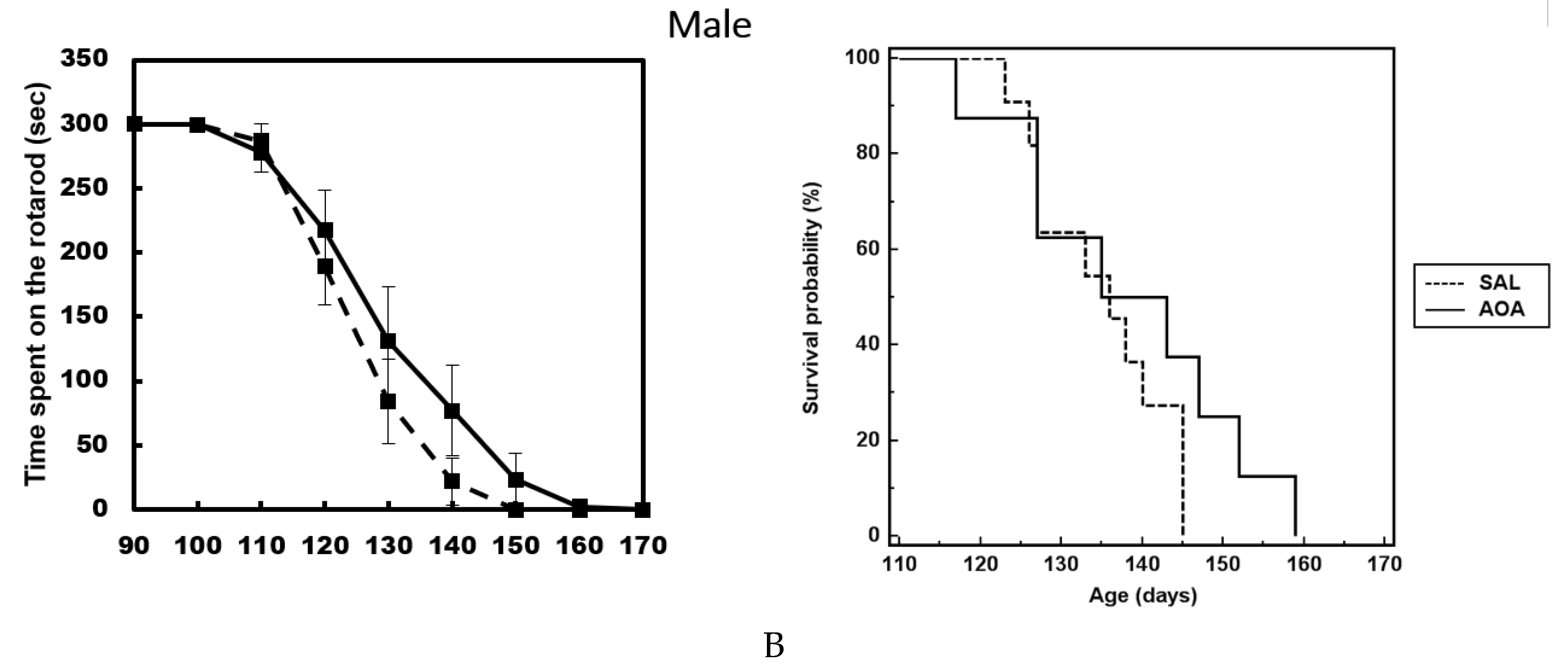
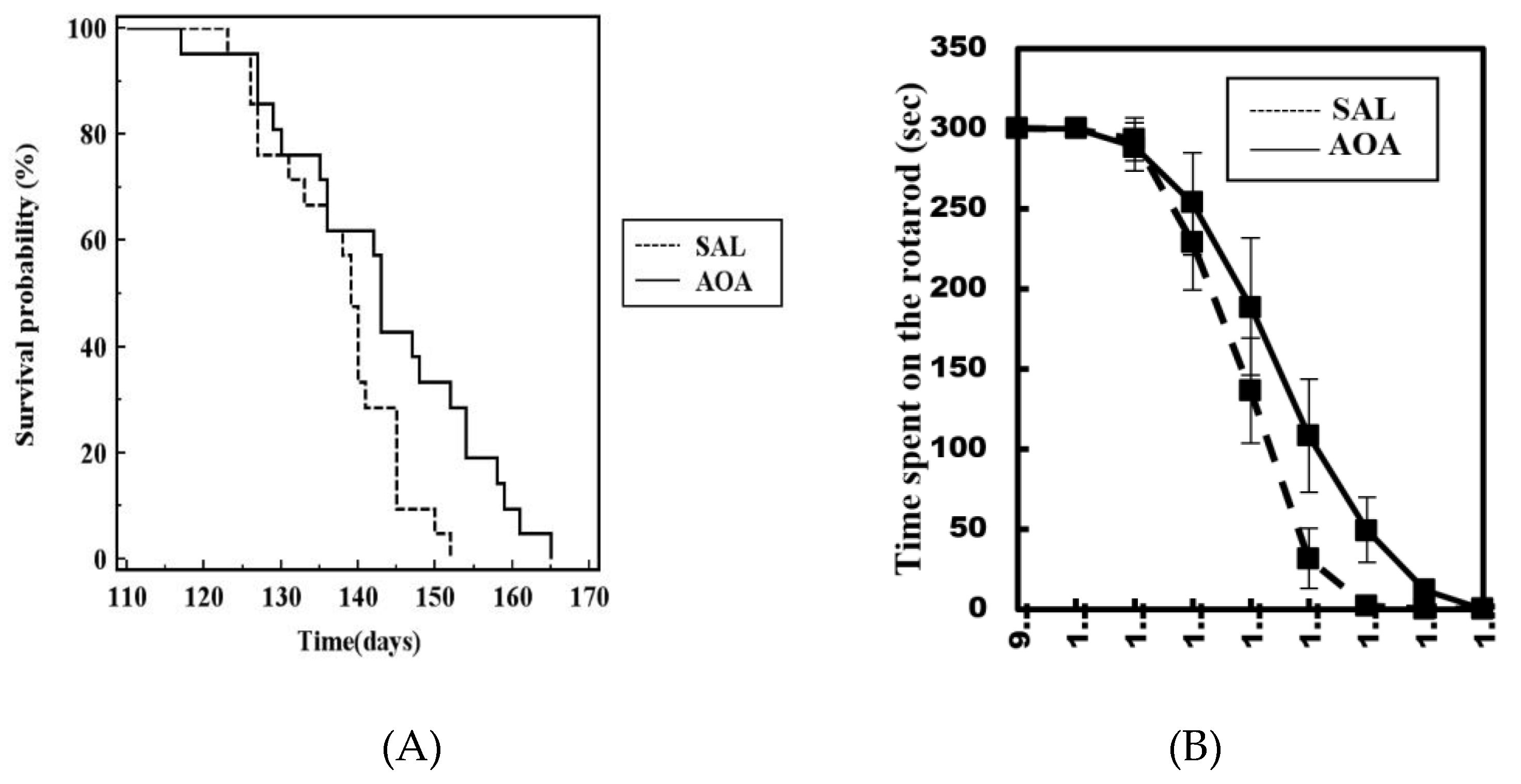
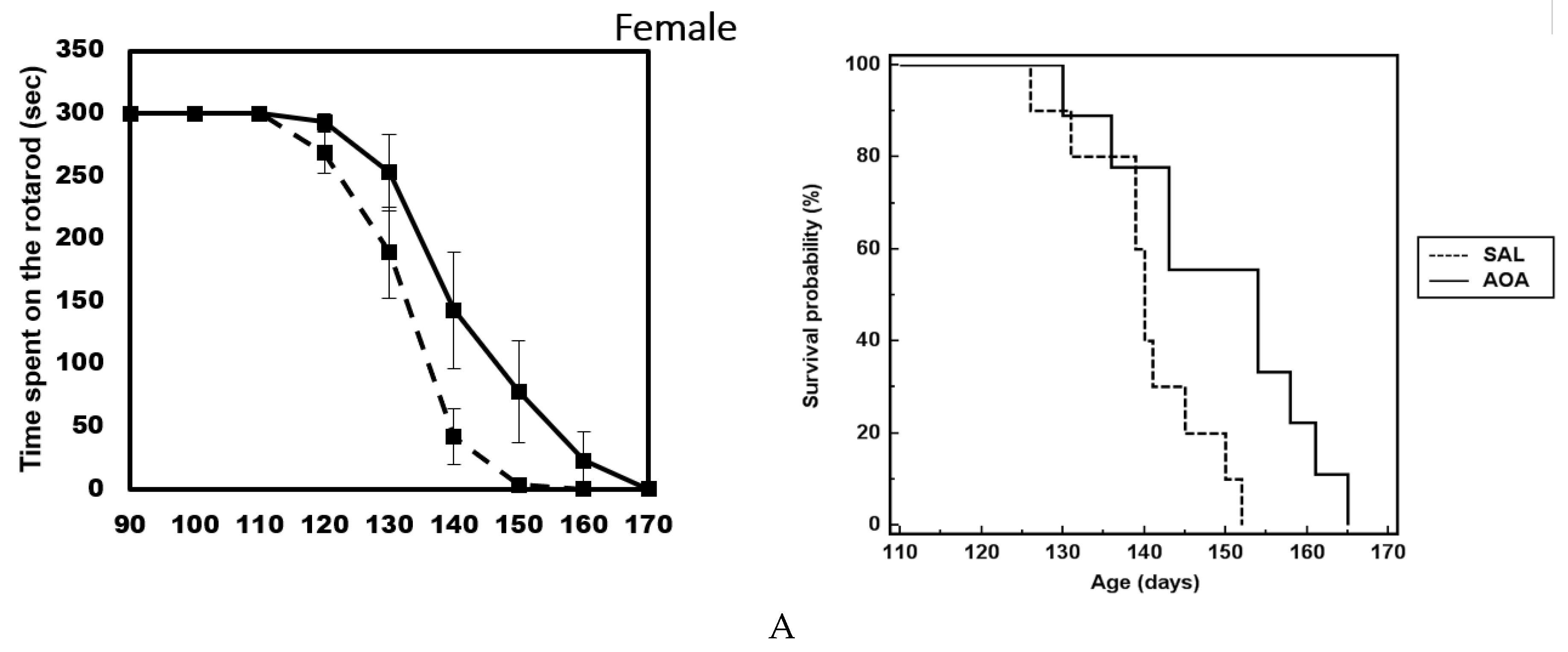
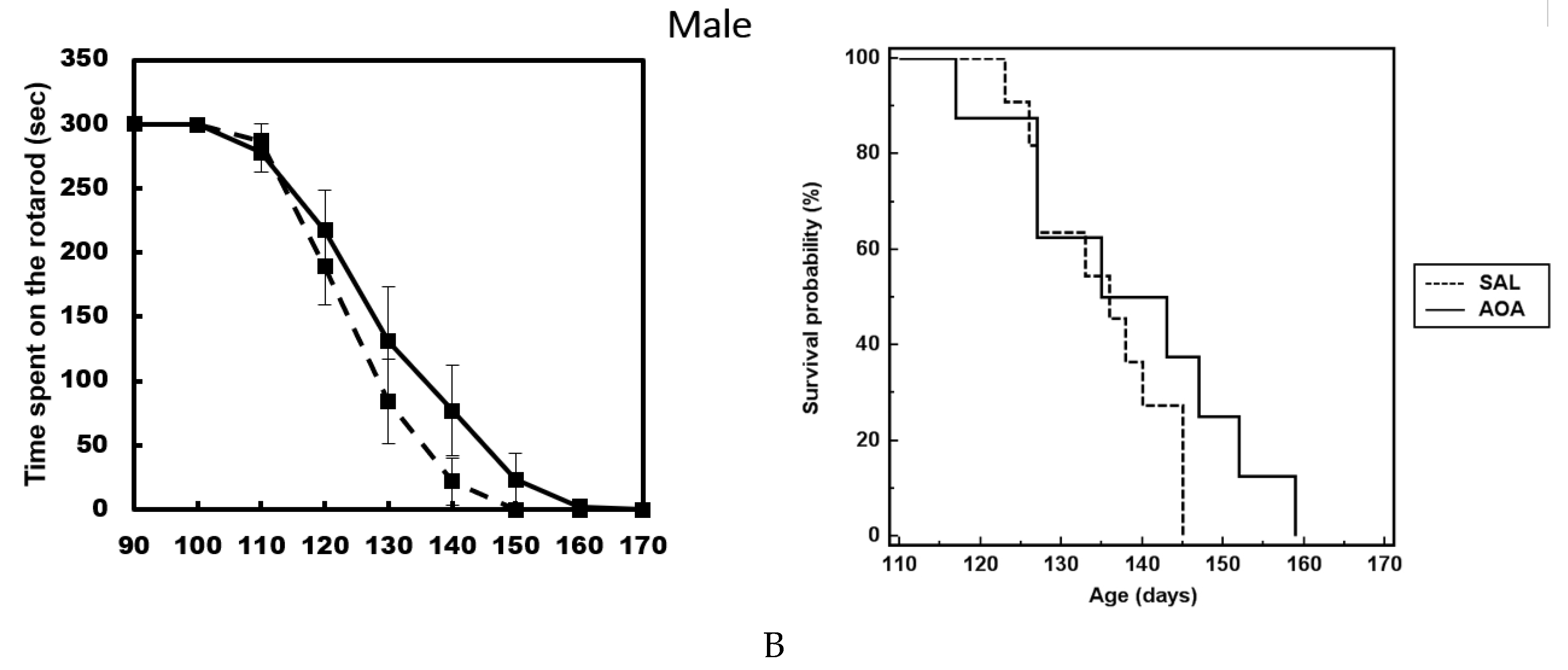
2.3. Effect of AOA Treatment on Motor Dysfunction, Body Weight and Survival of SOD1G93A Mice
2.4. AOA Treatment Decreases the Amount of H2S in the Cerebral Tissues
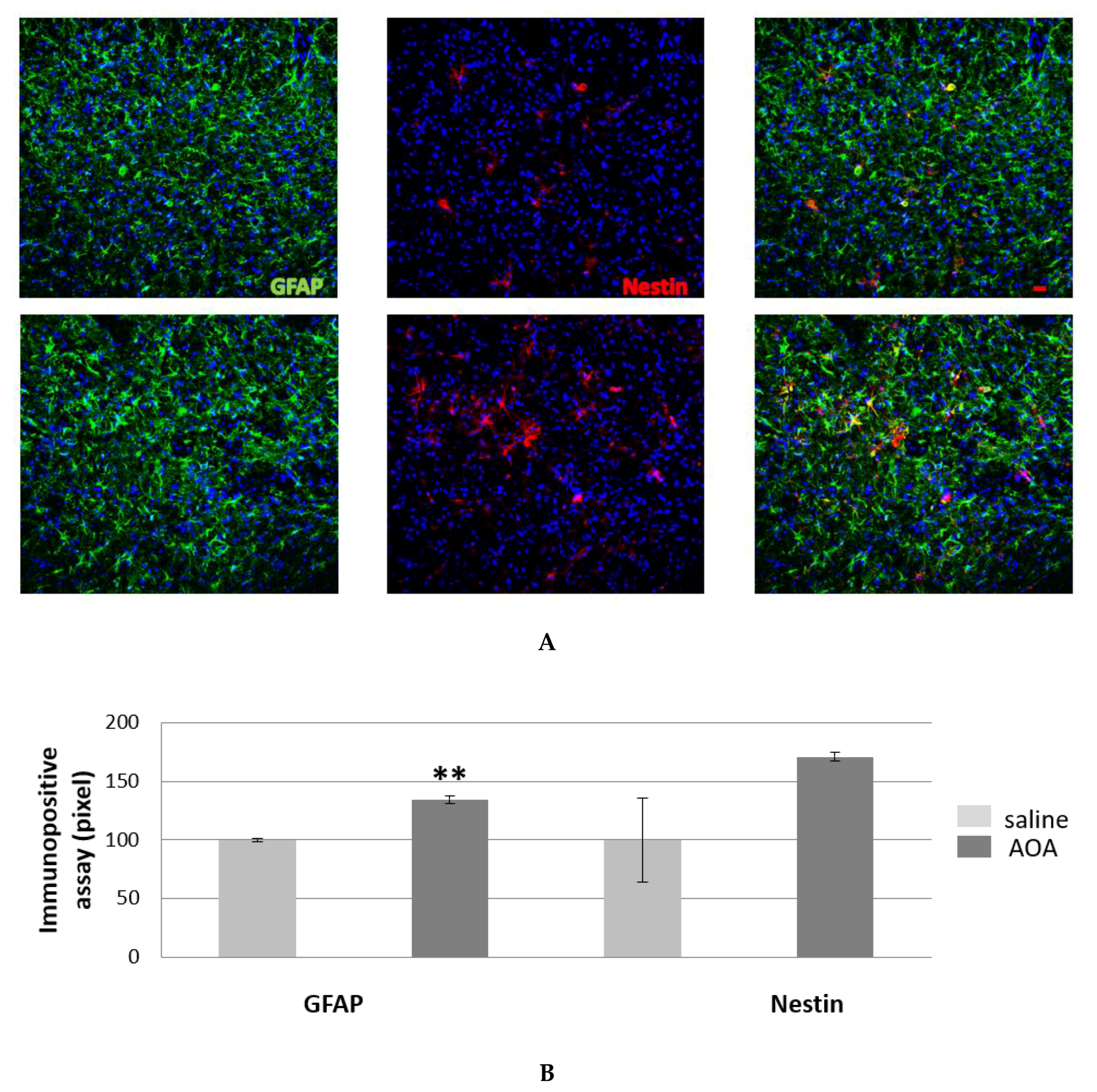
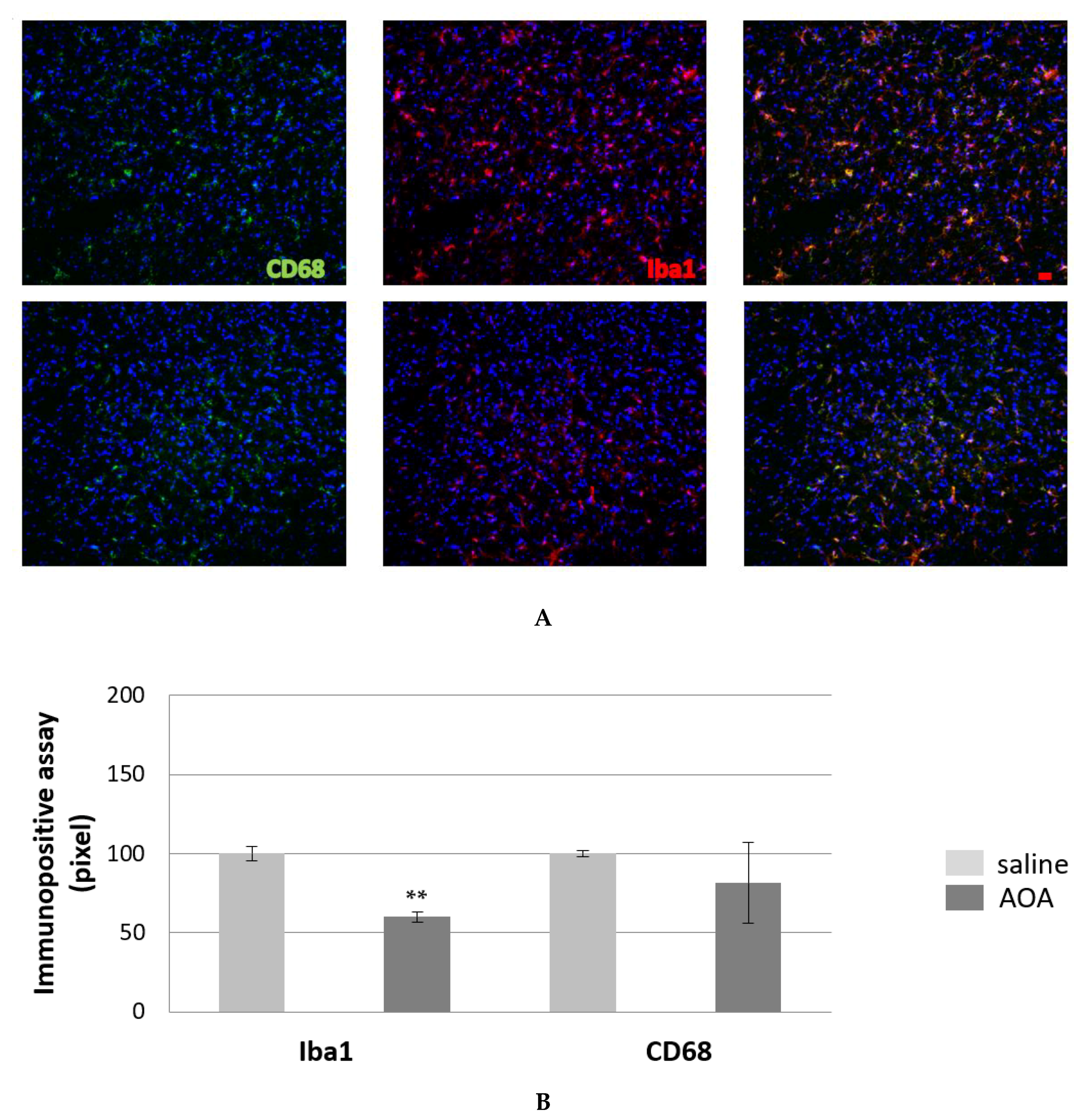
2.5. Effect of AOA on Spinal cord Glial Cells
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Mixed Spinal Cord Cultures
4.3. Drug Treatment and Disease Progression Assessment
4.4. Whole Tissue Preparation for Metabolomics Analyses and Immunofluorescence Procedures
4.5. Immunocytochemistry
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Turner, M.R.; Swash, M. The expanding syndrome of amyotrophic lateral sclerosis: A clinical and molecular odyssey. J. Neurol. Neurosurg. Psychiatry 2015, 86, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Robberecht, W. The phenotypic variability of amyotrophic lateral sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Anagnostou, G.; Chai, A.; Withers, J.; Morris, A.; Adhikaree, J.; Pennetta, G.; de Belleroche, J.S. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 40266–40281. [Google Scholar] [CrossRef]
- Sapp, P.C.; Hosler, B.A.; McKenna-Yasek, D.; Chin, W.; Gann, A.; Genise, H.; Gorenstein, J.; Huang, M.; Sailer, W.; Scheffler, M.; et al. Identification of two novel loci for dominantly inherited familial amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2003, 73, 397–403. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Barbeito, A.G.; Mesci, P.; Boillée, S. Motor neuron-immune interactions: The vicious circle of ALS. J. Neural Transm. (Vienna) 2010, 117, 981–1000. [Google Scholar] [CrossRef]
- Puentes, F.; Malaspina, A.; van Noort, J.M.; Amor, S. Non-neuronal Cells in ALS: Role of Glial, Immune cells and Blood-CNS Barriers. Brain Pathol. 2016, 26, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Moisse, K.; Strong, M.J. Innate immunity in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1083–1093. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef]
- Davoli, A.; Greco, V.; Spalloni, A.; Guatteo, E.; Neri, C.; Rizzo, G.R.; Cordella, A.; Romigi, A.; Cortese, C.; Bernardini, S.; et al. Evidence of hydrogen sulfide involvement in amyotrophic lateral sclerosis. Ann. Neurol. 2015, 77, 697–709. [Google Scholar] [CrossRef]
- Greco, V.; Spalloni, A.; Corasolla Carregari, V.; Pieroni, L.; Persichilli, S.; Mercuri, N.B.; Urbani, A.; Longone, P. Proteomics and Toxicity Analysis of Spinal-Cord Primary Cultures upon Hydrogen Sulfide Treatment. Antioxidants 2018, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Niu, Y.Y.; Jiang, W.Z.; Tang, H.L.; Zhang, C.; Xia, Q.M.; Tang, X.Q. Neuroprotective effects of hydrogen sulfide and the underlying signaling pathways. Rev. Neurosci. 2015, 26, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Pagliusi, S.R.; Gomes, C.; Leite, J.R.; Trolin, G. Aminooxyacetic acid induced accumulation of GABA in the rat brain. Interaction with GABA receptors and distribution in compartments. Naunyn Schmiedebergs Arch. Pharmacol. 1983, 322, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Whiteman, M.; Cirino, G. Pharmacological tools for hydrogen sulphide research: A brief, introductory guide for beginners. Br. J. Pharmacol. 2015, 172, 1633–1637. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Khan, A.H.; Islam, M.T.; Prieto, M.C.; Majid, D.S. Interdependency of cystathione γ-lyase and cystathione β-synthase in hydrogen sulfide-induced blood pressure regulation in rats. Am. J. Hypertens. 2012, 25, 74–81. [Google Scholar] [CrossRef]
- Veldink, J.H.; Bär, P.R.; Joosten, E.A.; Otten, M.; Wokke, J.H.; van den Berg, L.H. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul. Disord. 2003, 9, 737–743. [Google Scholar] [CrossRef]
- McCombe, P.A.; Henderson, R.D. Effects of gender in amyotrophic lateral sclerosis. Gend. Med. 2010, 7, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Wiliński, B.; Wiliński, J.; Somogyi, E.; Piotrowska, J.; Góralska, M. Digoxin increases hydrogen sulfide concentrations in brain, heart and kidney tissues in mice. Pharmacol. Rep. 2011, 63, 1243–1247. [Google Scholar] [PubMed]
- Wiliński, B.; Wiliński, J.; Somogyi, E.; Piotrowska, J.; Opoka, W. Metformin raises hydrogen sulfide tissue concentrations in various mouse organs. Pharmacol. Rep. 2013, 65, 737–742. [Google Scholar] [CrossRef]
- Lee, M.; Schwab, C.; Yu, S.; McGeer, E.; McGeer, P.L. Astrocytes produce the antiinflammatory and neuroprotective agent hydrogen sulfide. Neurobiol. Aging 2009, 30, 1523–1534. [Google Scholar] [CrossRef]
- Enokido, Y.; Suzuki, E.; Iwasawa, K.; Namekata, K.; Okazawa, H.; Kimura, H. Cystathionine β-synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J. 2005, 19, 1854–1856. [Google Scholar] [CrossRef]
- Frederiksen, K.; McKay, R.D. Proliferation and differentiation of rat neuroepithelial precursor cells in vivo. J. Neurosci. 1988, 8, 1144–1151. [Google Scholar] [CrossRef] [Green Version]
- Fiedorowicz, A.; Figiel, I.; Kamińska, B.; Zaremba, M.; Wilk, S.; Oderfeld-Nowak, B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res. 2001, 912, 116–127. [Google Scholar] [CrossRef]
- Lendahl, U.; Zimmerman, L.B.; McKay, R.D. CNS stem cells express a new class of intermediate filament protein. Cell 1990, 60, 585–595. [Google Scholar] [CrossRef]
- Bonaguidi, M.A.; Wheeler, M.A.; Shapiro, J.S.; Stadel, R.P.; Sun, G.J.; Ming, G.L.; Song, H. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 2011, 145, 1142–1155. [Google Scholar] [CrossRef]
- Namiki, J.; Tator, C.H. Cell proliferation and nestin expression in the ependyma of the adult rat spinal cord after injury. J. Neuropathol. Exp. Neurol. 1999, 58, 489–498. [Google Scholar] [CrossRef]
- Wiese, C.; Rolletschek, A.; Kania, G.; Blyszczuk, P.; Tarasov, K.V.; Tarasova, Y.; Wersto, R.P.; Boheler, K.R.; Wobus, A.M. Nestin expression—A property of multi-lineage progenitor cells? Cell. Mol. Life Sci. 2004, 61, 2510–2522. [Google Scholar] [CrossRef]
- Selvaraj, V.; Plane, J.M.; Williams, A.J.; Deng, W. Switching cell fate: The remarkable rise of induced pluripotent stem cells and lineage reprogramming technologies. Trends Biotechnol. 2010, 28, 214–223. [Google Scholar] [CrossRef]
- Buffo, A.; Rolando, C.; Ceruti, S. Astrocytes in the damaged brain: Molecular and cellular insights into their reactive response and healing potential. Biochem. Pharmacol. 2010, 79, 77–89. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Ibata, I.; Ito, D.; Ohsawa, K.; Kohsaka, S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem. Biophys. Res. Commun. 1996, 224, 855–862. [Google Scholar] [CrossRef]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Slepko, N.; Levi, G. Progressive activation of adult microglial cells in vitro. Glia 1996, 16, 241–246. [Google Scholar] [CrossRef]
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharideinduced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [CrossRef]
- Zhang, J.; Sio, S.W.S.; Moochhala, S.; Bhatia, M. Role of hydrogen sulfide in severe burn injury-induced inflammation in mice. Mol. Med. 2010, 16, 417–424. [Google Scholar] [CrossRef]
- Ekundi-Valentim, E.; Santos, K.T.; Camargo, E.A.; Denadai-Souza, A.; Teixeira, S.A.; Zanoni, C.I.; Grant, A.D.; Wallace, J.; Muscará, M.N.; Costa, S.K. Differing effects of exogenous and endogenous hydrogen sulphide in carrageenan-induced knee joint synovitis in the rat. Br. J. Pharmacol. 2010, 159, 1463–1474. [Google Scholar] [CrossRef] [Green Version]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef]
- Fiorucci, S.; Orlandi, S.; Mencarelli, A.; Caliendo, G.; Santagada, V.; Distrutti, E.; Santucci, L.; Cirino, G.; Wallace, J.L. Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br. J. Pharmacol. 2007, 150, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Kimura, H. Hydrogen sulfide protects neurons from oxidative stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.; Kimura, H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Espey, M.G.; Miranda, K.M.; Feelisch, M.; Fukuto, J.; Grisham, M.B.; Vitek, M.P.; Wink, D.A. Mechanisms of cell death governed by the balance between nitrosative and oxidative stress. Ann. N. Y. Acad. Sci. 2000, 899, 209–221. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Carbon monoxide: Present and future indications for a medical gas. Korean J. Intern. Med. 2013, 28, 123–140. [Google Scholar] [CrossRef]
- He, X.L.; Yan, N.; Chen, X.S.; Qi, Y.W.; Yan, Y.; Cai, Z. Hydrogen sulfide down-regulates BACE1 and PS1 via activating PI3K/Akt pathway in the brain of APP/PS1 transgenic mouse. Pharmacol. Rep. 2016, 68, 975–982. [Google Scholar] [CrossRef]
- Cao, X.; Cao, L.; Ding, L.; Bian, J.S. A new hope for a devastating disease: Hydrogen sulfide in Parkinson’s disease. Mol. Neurobiol. 2017, 55, 3789–3799. [Google Scholar] [CrossRef]
- Beery, A.K.; Zucker, I. Sex bias in neuroscience and biomedical research. Neurosci. Biobehav. Rev. 2011, 35, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Haverkamp, L.J.; Appel, V.; Appel, S.H. Natural history of amyotrophic lateral sclerosis in a database population. Validation of a scoring system and a model for survival prediction. Brain 1995, 118, 707–719. [Google Scholar] [CrossRef]
- Nefussy, B.; Drory, V.E. Moving toward a predictive and personalized clinical approach in amyotrophic lateral sclerosis: Novel developments and future directions in diagnosis, genetics, pathogenesis and therapies. EPMA J. 2010, 1, 329–341. [Google Scholar] [CrossRef]
- Beghi, E.; Logroscino, G.; Chiò, A.; Hardiman, O.; Millul, A.; Mitchell, D.; Swingler, R.; Traynor, B.J. Amyotrophic lateral sclerosis, physical exercise, trauma and sports: Results of a population-based pilot case-control study. Amyotroph. Lateral Scler. 2010, 11, 289–292. [Google Scholar] [CrossRef]
- De Jong, S.; Huisman, M.; Sutedja, N.; van der Kooi, A.; de Visser, M.; Schelhaas, J.; van der Schouw, Y.; Veldink, J.; van den Berg, L. Endogenous female reproductive hormones and the risk of amyotrophic lateral sclerosis. J. Neurol. 2013, 260, 507–512. [Google Scholar] [CrossRef]
- Chio, A.; Meineri, P.; Tribolo, A.; Schiffer, D. Risk factors in motorneuron disease: A case-control study. Neuroepidemiology 1991, 10, 174–184. [Google Scholar] [CrossRef]
- D’Alessandro, A.; D’Aguanno, S.; Cencioni, M.T.; Pieroni, L.; Diamantini, A.; Battistini, L.; Longone, P.; Spalloni, A.; De Laurenzi, V.; Bernardini, S.; et al. Protein repertoire impact of Ubiquitin-Proteasome System impairment: Insight into the protective role of beta-estradiol. J. Proteom. 2012, 75, 1440–1453. [Google Scholar] [CrossRef]
- Heitzer, M.; Kaiser, S.; Kanagaratnam, M.; Zendedel, A.; Hartmann, P.; Beyer, C.; Johann, S. Administration of 17β-Estradiol Improves Motoneuron Survival and Down-regulates Inflammasome Activation in Male SOD1(G93A) ALS Mice. Mol. Neurobiol. 2017, 54, 8429–8443. [Google Scholar] [CrossRef]
- Lechuga, T.J.; Bilg, A.K.; Patel, B.A.; Nguyen, N.A.; Qi, Q.R.; Chen, D.B. Estradiol-17β stimulates H(2) S biosynthesis by ER-dependent CBS and CSE transcription in uterine artery smooth muscle cells in vitro. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef]
- Bucci, M.; Mirone, V.; Di Lorenzo, A.; Vellecco, V.; Roviezzo, F.; Brancaleone, V.; Ciro, I.; Cirino, G. Hydrogen sulphide is involved in testosterone vascular effect. Eur. Urol. 2009, 56, 378–383. [Google Scholar] [CrossRef]
- Brancaleone, V.; Vellecco, V.; Matassa, D.S.; d’Emmanuele di Villa Bianca, R.; Sorrentino, R.; Ianaro, A.; Bucci, M.; Esposito, F.; Cirino, G. Crucial role of androgen receptor in vascular H2S biosynthesis induced by testosterone. Br. J. Pharmacol. 2015, 172, 1505–1515. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Spalloni, A.; Geracitano, R.; Berretta, N.; Sgobio, C.; Bernardi, G.; Mercuri, N.B.; Longone, P.; Ammassari-Teule, M. Molecular and synaptic changes in the hippocampus underlying superior spatial abilities in pre-symptomatic G93A+/+ mice overexpressing the human Cu/Zn superoxide dismutase (Gly93 → ALA) mutation. Exp. Neurol. 2006, 197, 505–514. [Google Scholar] [CrossRef]
- Spalloni, A.; Albo, F.; Ferrari, F.; Mercuri, N.; Bernardi, G.; Zona, C.; Longone, P. Cu/Zn-superoxide dismutase (GLY93 --> ALA) mutation alters AMPA receptor subunit expression and function and potentiates kainate-mediated toxicity in motor neurons in culture. Neurobiol. Dis. 2004, 2, 340–350. [Google Scholar] [CrossRef]
- Bailey, B.; Waraska, J.; Acworth, I. Direct determination of tissue aminothiol, disulfide, and thioether levels using HPLC-ECD with a novel stable boron-doped diamond working electrode. Methods Mol. Biol. 2010, 594, 327–339. [Google Scholar]
- Gallaher, Z.R.; Ryu, V.; Herzog, T.; Ritter, R.C.; Czaja, K. Changes in microglial activation within the hindbrain, nodose ganglia, and the spinal cord following subdiaphragmatic vagotomy. Neurosci. Lett. 2012, 513, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Nutini, M.; Spalloni, A.; Florenzano, F.; Westenbroek, R.E.; Marini, C.; Catterall, W.A.; Bernardi, G.; Longone, P. Increased expression of the beta3 subunit of voltage-gated Na+ channels in the spinal cord of the SOD1G93A mouse. Mol. Cell. Neurosci. 2011, 47, 108–118. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2S mg/L | ALS Total n = 37 | |
| Female (n = 16) | Male (n = 21) | |
| 5.52 | 6.38 | |
| 7.54 | 4.49 | |
| 13.29 | 4.60 | |
| 13.89 | 3.18 | |
| 8.44 | 3.49 | |
| 5.32 | 7.54 | |
| 4.38 | 6.60 | |
| 8.05 | 6.02 | |
| 3.86 | 5.04 | |
| 9.25 | 4.01 | |
| 7.81 | 9.25 | |
| 13.40 | 7.64 | |
| 8.75 | 11.51 | |
| 10.42 | 12.39 | |
| 2.66 | 8.41 | |
| 13.34 | 10.43 | |
| 10.35 | ||
| 11.30 | ||
| 5.19 | ||
| 6.77 | ||
| 3.12 | ||
| Mean value | 8.49 ± 3.62 | 7.03 ± 2.92 |
| H2S (MG/L) | Days in Culture | ||||
|---|---|---|---|---|---|
| II | IV | V | VI | VII | |
| NT (Not Treated) | 4.3 ± 0.4 | 4.5 ± 0.03 | 3.8 ± 0.26 | 3.2 ± 0.14 | 1.42 ± 0.16 |
| 250 µM AOA | 2.7 ± 0.2 | 2 ± 0.02 | 1.4 ± 0.015 | 0.7 ± 0.21 * | ND |
| 500 µM AOA | 1.8 ± 0.25 * | 0.9 ± 0.08 * | ND | ND | ND |
| 1 MM AOA | 0.4 ± 0.26 * | ND | ND | ND | ND |
| 2 MM AOA | ND | ND | ND | ND | ND |
| H2S (mg/L) | G93A | |||
|---|---|---|---|---|
| Male | Male AOA | FEMALE | Female AOA | |
| Spinal Cord | 4.09 ± 0.06 *** | 1.41 ±0.03 | 5.29 ± 0.26 *** | 1.37 ± 0.021 |
| Brainstem | 3.71 ± 0.2 *** | 1.31 ±0.02 | 4.00 ± 0.015 *** | 1.56 ± 0.014 |
| Motor Cortex | 3.99 ± 0.08 *** | 1.4 ± 0.15 | 5.00 ± 0.07 *** | 1.87 ± 0.017 |
| Muscle | 3.60 ± 0.031 | 2.83 ± 0.04 | 4.02 ± 0.12 | 3.91 ± 0.012 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spalloni, A.; Greco, V.; Ciriminna, G.; Corasolla Carregari, V.; Marini, F.; Pieroni, L.; Mercuri, N.B.; Urbani, A.; Longone, P. Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model. Int. J. Mol. Sci. 2019, 20, 2550. https://doi.org/10.3390/ijms20102550
Spalloni A, Greco V, Ciriminna G, Corasolla Carregari V, Marini F, Pieroni L, Mercuri NB, Urbani A, Longone P. Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model. International Journal of Molecular Sciences. 2019; 20(10):2550. https://doi.org/10.3390/ijms20102550
Chicago/Turabian StyleSpalloni, Alida, Viviana Greco, Giulia Ciriminna, Victor Corasolla Carregari, Federica Marini, Luisa Pieroni, Nicola B. Mercuri, Andrea Urbani, and Patrizia Longone. 2019. "Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model" International Journal of Molecular Sciences 20, no. 10: 2550. https://doi.org/10.3390/ijms20102550
APA StyleSpalloni, A., Greco, V., Ciriminna, G., Corasolla Carregari, V., Marini, F., Pieroni, L., Mercuri, N. B., Urbani, A., & Longone, P. (2019). Impact of Pharmacological Inhibition of Hydrogen Sulphide Production in the SOD1G93A-ALS Mouse Model. International Journal of Molecular Sciences, 20(10), 2550. https://doi.org/10.3390/ijms20102550