Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor H (CFH) Antibody Ab42 and CFH Polypeptide


 and
and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. The Three-Dimensional (3D) Structure of Ab42
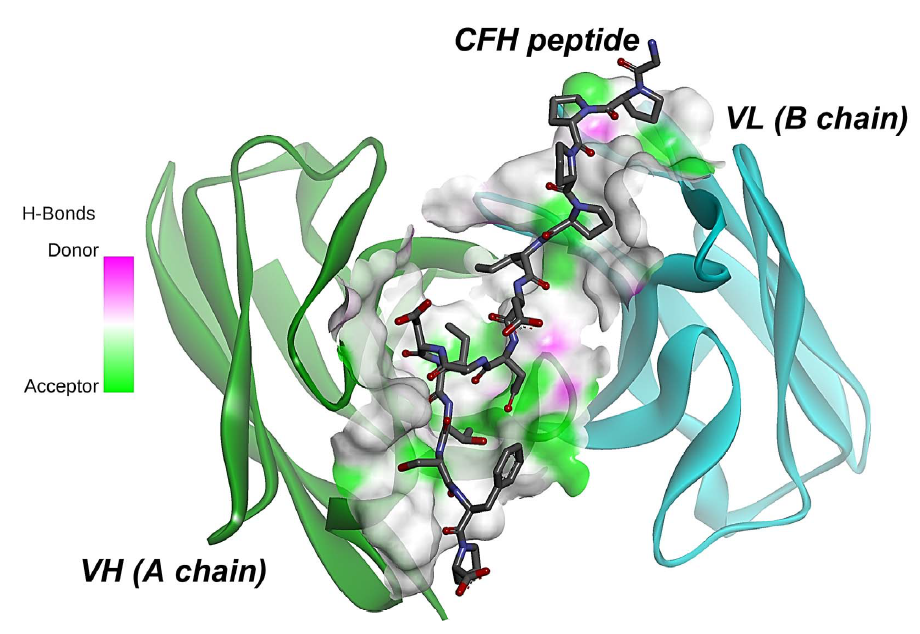
2.2. Molecular Docking
2.3. Stability Analysis of Ab42-pCFH Complex
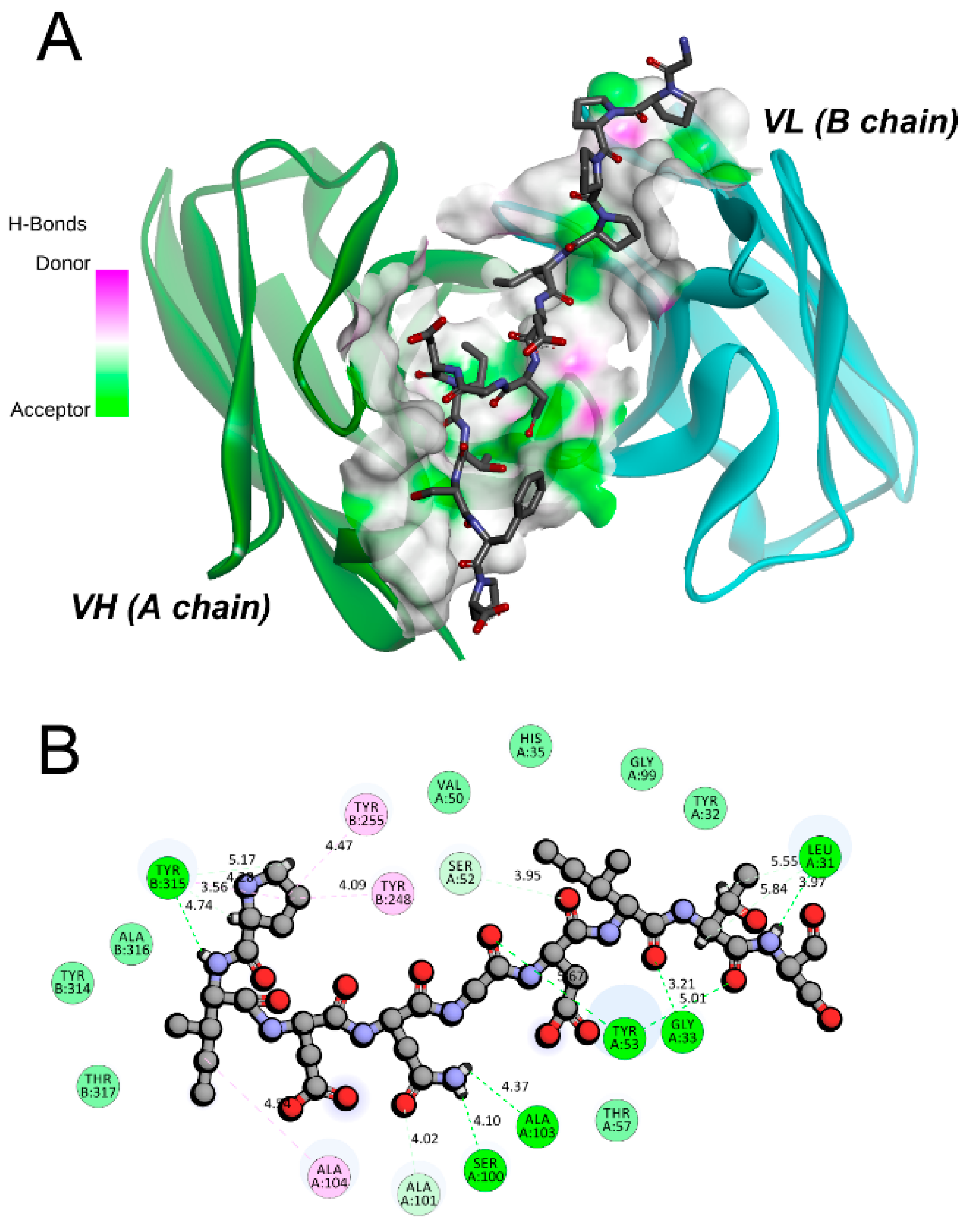
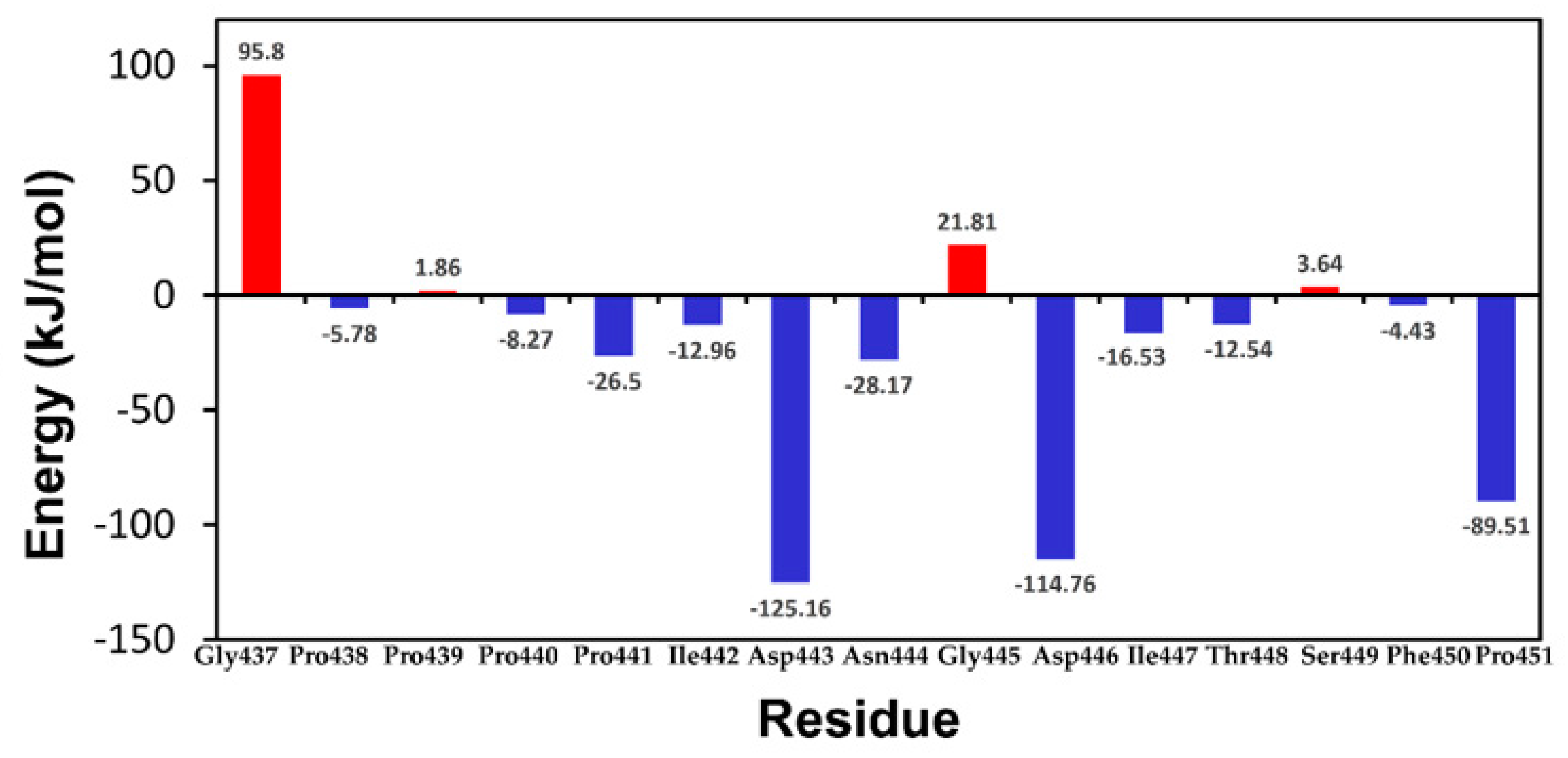
2.4. MM-PBSA and Energy Decomposition
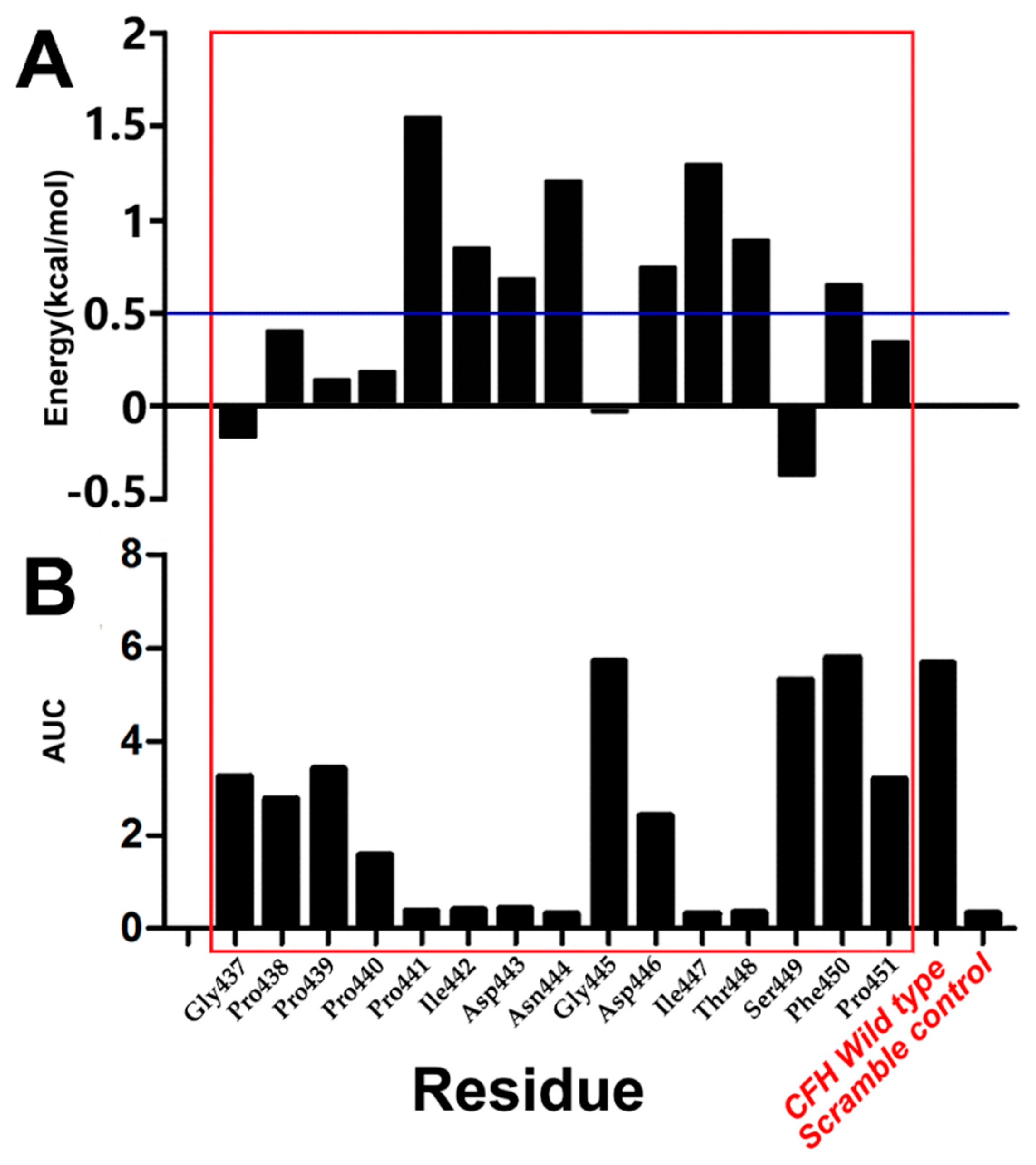
2.5. Comparison of CAS and EAS
2.6. Distance Monitoring of Key Amino Acids
3. Materials and Methods
3.1. Experimental Preparation
3.2. Molecular Docking
3.3. MD Simulations
3.4. Binding Energy Calculation by MM-PBSA
3.5. CAS
3.6. EAS
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bushey, R.T.; Moody, M.A.; Nicely, N.L.; Haynes, B.F.; Alam, S.M.; Keir, S.T.; Bentley, R.C.; Roy Choudhury, K.; Gottlin, E.B.; Campa, M.J.; et al. A Therapeutic Antibody for Cancer, Derived from Single Human B Cells. Cell Rep. 2016, 15, 1505–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbert, A.P.; Kavanagh, D.; Johansson, C.; Morgan, H.P.; Blaum, B.S.; Hannan, J.P.; Barlow, P.N.; Uhrin, D. Structural and functional characterization of the product of disease-related factor H gene conversion. Biochemistry 2012, 51, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Amornsiripanitch, N.; Hong, S.; Campa, M.J.; Frank, M.M.; Gottlin, E.B.; Patz, E.F., Jr. Complement factor H autoantibodies are associated with early stage NSCLC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 3226–3231. [Google Scholar] [CrossRef] [PubMed]
- Kajander, T.; Lehtinen, M.J.; Hyvarinen, S.; Bhattacharjee, A.; Leung, E.; Isenman, D.E.; Meri, S.; Goldman, A.; Jokiranta, T.S. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. USA 2011, 108, 2897–2902. [Google Scholar] [CrossRef]
- Ferreira, V.P.; Pangburn, M.K.; Cortes, C. Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Campa, M.J.; Gottlin, E.B.; Bushey, R.T.; Patz, E.F., Jr. Complement Factor H Antibodies from Lung Cancer Patients Induce Complement-Dependent Lysis of Tumor Cells, Suggesting a Novel Immunotherapeutic Strategy. Cancer Immunol. Res. 2015, 3, 1325–1332. [Google Scholar] [CrossRef]
- Hofer, J.; Giner, T.; Jozsi, M. Complement factor H-antibody-associated hemolytic uremic syndrome: Pathogenesis, clinical presentation, and treatment. Semin. Thromb. Hemost. 2014, 40, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.X.; Levesque, M.C.; Nagel, A.; Dixon, A.; Zhang, R.; Walter, E.; Parks, R.; Whitesides, J.; Marshall, D.J.; Hwang, K.K.; et al. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J. Virol. Methods 2009, 158, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, M.; Boido, V.; Colla, P.; Loddo, R.; Posocco, P.; Paneni, M.S.; Fermeglia, M.; Pricl, S. Pharmacophore modeling, resistant mutant isolation, docking, and MM-PBSA analysis: Combined experimental/computer-assisted approaches to identify new inhibitors of the bovine viral diarrhea virus (BVDV). Bioorg. Med. Chem. 2010, 18, 2304–2316. [Google Scholar] [CrossRef]
- Luo, Q.; Zhang, C.; Miao, L.; Zhang, D.; Bai, Y.; Hou, C.; Liu, J.; Yan, F.; Mu, Y.; Luo, G. Triple mutated antibody scFv2F3 with high GPx activity: Insights from MD, docking, MDFE, and MM-PBSA simulation. Amino Acids 2013, 44, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Chen, C.F.; Zhao, B.B.; Gong, L.L.; Jin, W.J.; Liu, J.J.; Wang, J.F.; Wang, T.T.; Yuan, X.H.; He, Y.W. A novel antibody humanization method based on epitopes scanning and molecular dynamics simulation. PLoS ONE 2013, 8, e80636. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wu, X.; Yang, G.; Zu, Y.; Zhou, L. Understanding the Chiral Recognitions between Neuraminidases and Inhibitors: Studies with DFT, Docking and MD Methods. Int. J. Quantum Chem. 2011, 112, 909–921. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Showalter, S.A.; Bruschweiler, R. Validation of Molecular Dynamics Simulations of Biomolecules Using NMR Spin Relaxation as Benchmarks: Application to the AMBER99SB Force Field. J. Chem. Theory Comput. 2007, 3, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, C.; Zhang, J.Z.; Mei, Y. Evaluation of the Coupled Two-Dimensional Main Chain Torsional Potential in Modeling Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 267–274. [Google Scholar] [CrossRef]
- Jiayi, R.; Zhiwei, Y.; Nianjue, Z.; Junqi, L.; Chunlong, Y.; Shujian, L.; Bing, Y.; Junqing, H.; Huaxin, L.; Xiaohui, Y.; et al. Effect of ForceFields and Water Models of EGFRvIII (scFv) Complex by Molecular Dynamics Simulation, MM-PBSA Calculation, and ITC Experiment. Chem. J. Chin. Univ. 2017, 38, 2070–2076. [Google Scholar]
- Martins, S.A.; Perez, M.A.; Moreira, I.S.; Sousa, S.F.; Ramos, M.J.; Fernandes, P.A. Computational Alanine Scanning Mutagenesis: MM-PBSA vs TI. J. Chem. Theory Comput. 2013, 9, 1311–1319. [Google Scholar] [CrossRef]
- Liu, X.; Peng, L.; Zhou, Y.; Zhang, Y.; Zhang, J.Z.H. Computational Alanine Scanning with Interaction Entropy for Protein-Ligand Binding Free Energies. J. Chem. Theory Comput. 2018, 14, 1772–1780. [Google Scholar] [CrossRef]
- Gao, M.; Zhang, F.; Zhu, Y.; Gao, L.; Jiang, Y.; Luo, Y.; Zhuang, F.; Mao, Z.; Mao, J. Alanine scanning mutagenesis of SP70 epitope in characterizing speciesspecific antibodies induced by enterovirus 71based antigens. Mol. Med. Rep. 2018, 17, 1006–1014. [Google Scholar] [CrossRef]
- Yuan, X.; Qu, Z.; Wu, X.; Wang, Y.; Wei, F.; Zhang, H.; Liu, L.; Yang, Z. Homology Modeling and Evolution Trace Analysis of Human Adenovirus Type 3 Hexon. Chem. J Chin. Univ. 2009, 30, 1636–1640. [Google Scholar]
- Nicely, N.I.; Dennison, S.M.; Spicer, L.; Scearce, R.M.; Kelsoe, G.; Ueda, Y.; Chen, H.; Liao, H.X.; Alam, S.M.; Haynes, B.F. Crystal structure of a non-neutralizing antibody to the HIV-1 gp41 membrane-proximal external region. Nat. Struct. Mol. Biol. 2010, 17, 1492–1494. [Google Scholar] [CrossRef]
- Yuan, X.H.; Wang, Y.C.; Jin, W.J.; Zhao, B.B.; Chen, C.F.; Yang, J.; Wang, J.F.; Guo, Y.Y.; Liu, J.J.; Zhang, D.; et al. Structure-based high-throughput epitope analysis of hexon proteins in B and C species human adenoviruses (HAdVs). PLoS ONE 2012, 7, e32938. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, F.; Yuan, X.; Zhang, S. Molecular interactions of GABA analogues against the α+β-interface of GABAA receptor: Docking and molecular dynamics studies. Dig. J. Nanomater. Biostruct. 2015, 10, 811–822. [Google Scholar]
- Wiehe, K.; Pierce, B.; Tong, W.W.; Hwang, H.; Mintseris, J.; Weng, Z. The performance of ZDOCK and ZRANK in rounds 6-11 of CAPRI. Proteins 2007, 69, 719–725. [Google Scholar] [CrossRef]
- Wiehe, K.; Peterson, M.W.; Pierce, B.; Mintseris, J.; Weng, Z. Protein-protein docking: Overview and performance analysis. Methods Mol. Biol. 2008, 413, 283–314. [Google Scholar]
- Sapay, N.; Tieleman, D.P. Combination of the CHARMM27 force field with united-atom lipid force fields. J. Comput. Chem. 2011, 32, 1400–1410. [Google Scholar] [CrossRef]
- Agrawal, P.; Singh, H.; Srivastava, H.K.; Singh, S.; Kishore, G.; Raghava, G.P.S. Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinform. 2019, 19, 426. [Google Scholar] [CrossRef] [PubMed]
- Jalilzadeh-Razin, S.; Mantegi, M.; Tohidkia, M.R.; Pazhang, Y.; Pourseif, M.M.; Barar, J.; Omidi, Y. Phage antibody library screening for the selection of novel high-affinity human single-chain variable fragment against gastrin receptor: An in silico and in vitro study. Daru 2019. [Google Scholar] [CrossRef]
- Kumar, K.; Rajasekharan, S.; Gulati, S.; Rana, J.; Gabrani, R.; Jain, C.K.; Gupta, A.; Chaudhary, V.K.; Gupta, S. Elucidating the interacting domains of chandipura virus nucleocapsid protein. Adv. Virol. 2013, 2013, 594319. [Google Scholar] [CrossRef]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Proteins 2007, 67, 1078–1086. [Google Scholar] [CrossRef]
- Li, L.; Chen, R.; Weng, Z. RDOCK: Refinement of rigid-body protein docking predictions. Proteins 2003, 53, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Arthur, E.J.; Brooks, C.L., 3rd. Parallelization and improvements of the generalized born model with a simple sWitching function for modern graphics processors. J. Comput. Chem. 2016, 37, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Gerber, P.; Schulz-Gasch, T.; Stahl, M. Validation and use of the MM-PBSA approach for drug discovery. J. Med. Chem. 2005, 48, 4040–4048. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Yuan, X.H.; Wang, Y.C.; Qu, Z.Y.; Ren, J.Y.; Wu, X.M.; Wang, J.F. Phylogenetic and structural analysis of major surface proteins hemagglutinin and neuraminidase of novel avian influenza virus A H7N9 from chinese patient. Chem. Res. Chin. Uuiv. 2013, 29, 934–940. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Thompson, D.C.; Humblet, C.; Joseph-McCarthy, D. Investigation of MM-PBSA rescoring of docking poses. J. Chem. Inf. Model. 2008, 48, 1081–1091. [Google Scholar] [CrossRef]
- Li, T.; Froeyen, M.; Herdewijn, P. Computational alanine scanning and free energy decomposition for E. coli type I signal peptidase with lipopeptide inhibitor complex. J. Mol. Graph. Model. 2008, 26, 813–823. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor | Acceptor |
|---|---|
| Ab42-A:Gly33:HN | Pep-C:Ile447:O |
| Ab42-A:Tyr53:HH | Pep-C:Gly445:O |
| Ab42-A:Tyr53:HH | Pep-C:Gly448:O |
| Ab42-B:Tyr315:HH | Pep-C:Pro439:O |
| Pep-C:Ile442:HN | Ab42-B:Tyr315:O |
| Pep-C:Asn444:HD21 | Ab42-A:Ala103:O |
| Pep-C:Asn444:HD22 | Ab42-A:Ser100:O |
| Pep-C:Ser449:HN | Ab42-A:Leu31:O |
| Ab42-A:Gly33:HA2 | Pep-C:Ile447:O |
| Ab42-A:Ser52:HB1 | Pep-C:Asp446:O |
| Ab42-A:Ser52:HB2 | Pep-C:Asp446:O |
| Ab42-A:Ala101:HA | Pep-C:Asn444:OD1 |
| Pep-C:Pro441:HA | Ab42-B:Tyr315:O |
| Pep-C:Thr448:HA | Ab42-A:Leu31:O |
| Pep-C:Thr448:HB | Ab42-A:Leu31:O |
| Pep-C:Pro441:HD2 | Ab42-B:Tyr315:OH |
| CDR | a.a Position | Length (No. of a.a) | Sequence |
|---|---|---|---|
| HCDR1 | 26–33 | 8 | GFTFSLYG |
| HCDR2 | 51–58 | 8 | ISYDEKTK |
| HCDR3 | 97–107 | 12 | AKGSAEAATLDY |
| LCDR1 | 244–255 | 12 | QSLLYRSNKKNY |
| LCDR2 | 273–275 | 3 | WAS |
| LCDR3 | 312–320 | 9 | QQYYATPLT |
| Peptide ID | Mutation Position | Sequence |
|---|---|---|
| WT | N/A | GPPPPIDNGDITSFP |
| M1 | P437 | APPPPIDNGDITSFP |
| M2 | P438 | GAPPPIDNGDITSFP |
| M3 | P439 | GPAPPIDNGDITSFP |
| M4 | P440 | GPPAPIDNGDITSFP |
| M5 | P441 | GPPPAIDNGDITSFP |
| M6 | P442 | GPPPPADNGDITSFP |
| M7 | P443 | GPPPPIANGDITSFP |
| M8 | P444 | GPPPPIDAGDITSFP |
| M9 | P445 | GPPPPIDNADITSFP |
| M10 | P446 | GPPPPIDNGAITSFP |
| M11 | P447 | GPPPPIDNGDATSFP |
| M12 | P448 | GPPPPIDNGDIASFP |
| M13 | P449 | GPPPPIDNGDITAFP |
| M14 | P450 | GPPPPIDNGDITSAP |
| M15 | P451 | GPPPPIDNGDITSFA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Lin, S.-J.; Ren, J.-Y.; Liu, T.; Wang, Y.-M.; Li, C.-M.; Xu, W.-W.; He, Y.-W.; Zheng, W.-H.; Zhao, J.; et al. Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor H (CFH) Antibody Ab42 and CFH Polypeptide. Int. J. Mol. Sci. 2019, 20, 2568. https://doi.org/10.3390/ijms20102568
Yang B, Lin S-J, Ren J-Y, Liu T, Wang Y-M, Li C-M, Xu W-W, He Y-W, Zheng W-H, Zhao J, et al. Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor H (CFH) Antibody Ab42 and CFH Polypeptide. International Journal of Molecular Sciences. 2019; 20(10):2568. https://doi.org/10.3390/ijms20102568
Chicago/Turabian StyleYang, Bing, Shu-Jian Lin, Jia-Yi Ren, Tong Liu, Yue-Ming Wang, Cheng-Ming Li, Wen-Wen Xu, You-Wen He, Wei-Hong Zheng, Jian Zhao, and et al. 2019. "Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor H (CFH) Antibody Ab42 and CFH Polypeptide" International Journal of Molecular Sciences 20, no. 10: 2568. https://doi.org/10.3390/ijms20102568
APA StyleYang, B., Lin, S.-J., Ren, J.-Y., Liu, T., Wang, Y.-M., Li, C.-M., Xu, W.-W., He, Y.-W., Zheng, W.-H., Zhao, J., Yuan, X.-H., & Liao, H.-X. (2019). Molecular Docking and Molecular Dynamics (MD) Simulation of Human Anti-Complement Factor H (CFH) Antibody Ab42 and CFH Polypeptide. International Journal of Molecular Sciences, 20(10), 2568. https://doi.org/10.3390/ijms20102568