Coupling Genome-wide Transcriptomics and Developmental Toxicity Profiles in Zebrafish to Characterize Polycyclic Aromatic Hydrocarbon (PAH) Hazard


 ,
, 
Abstract
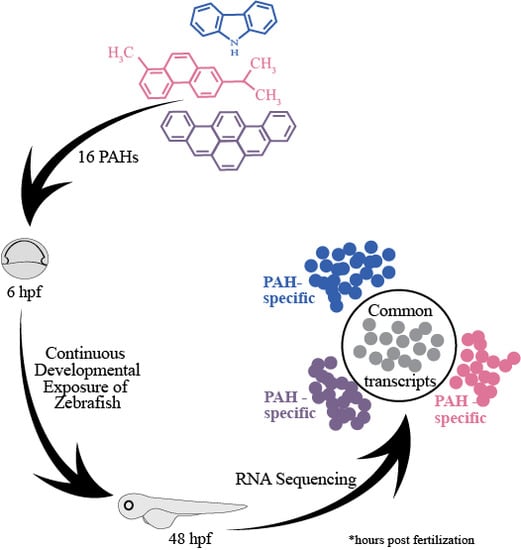
1. Introduction
2. Results and Discussion
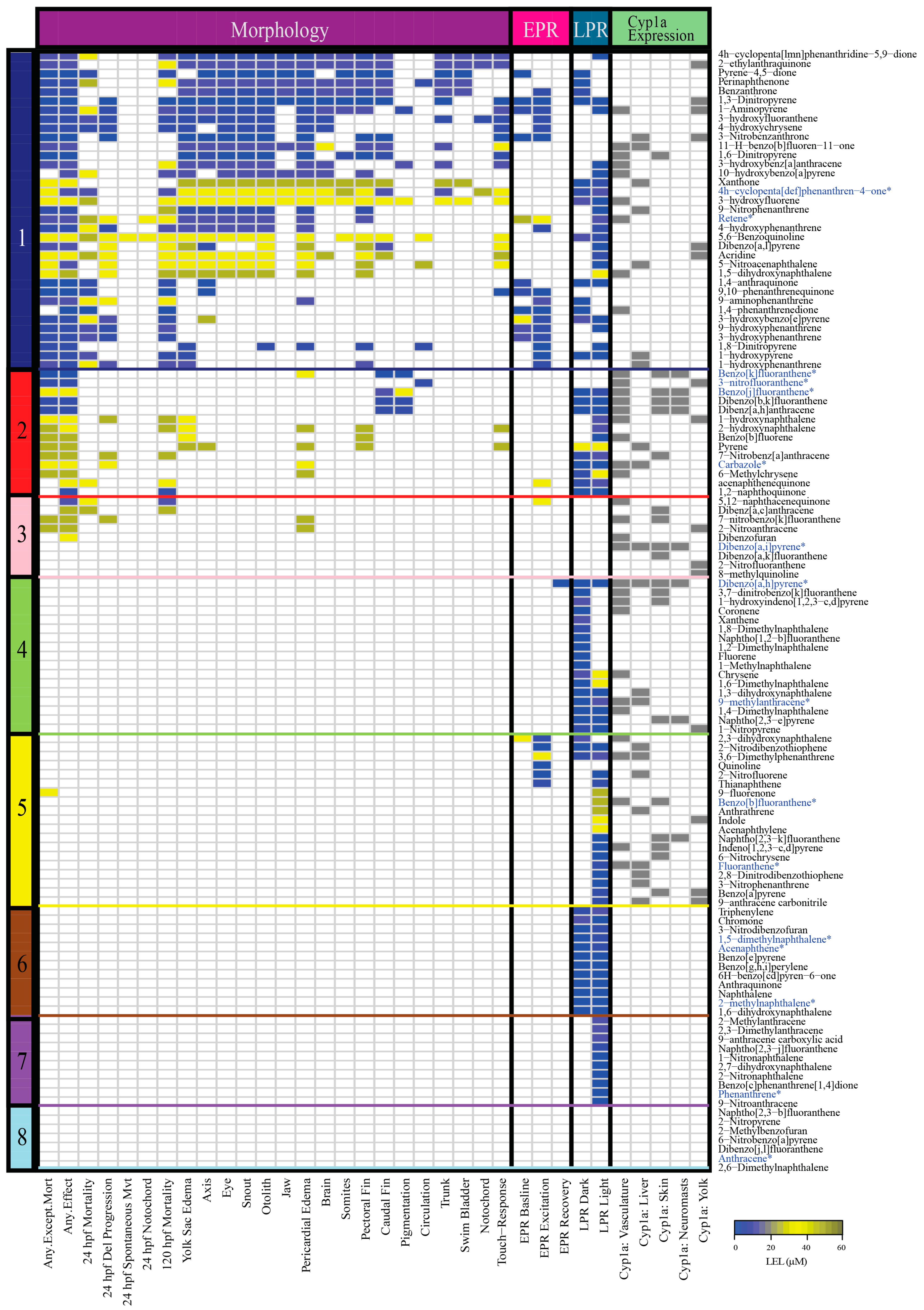
2.1. Classification of 123 PAHs into Eight Bins and Characterization of Bins
2.2. Selection of 16 Representative PAHs for RNA Sequencing from the Eight PAH Bins
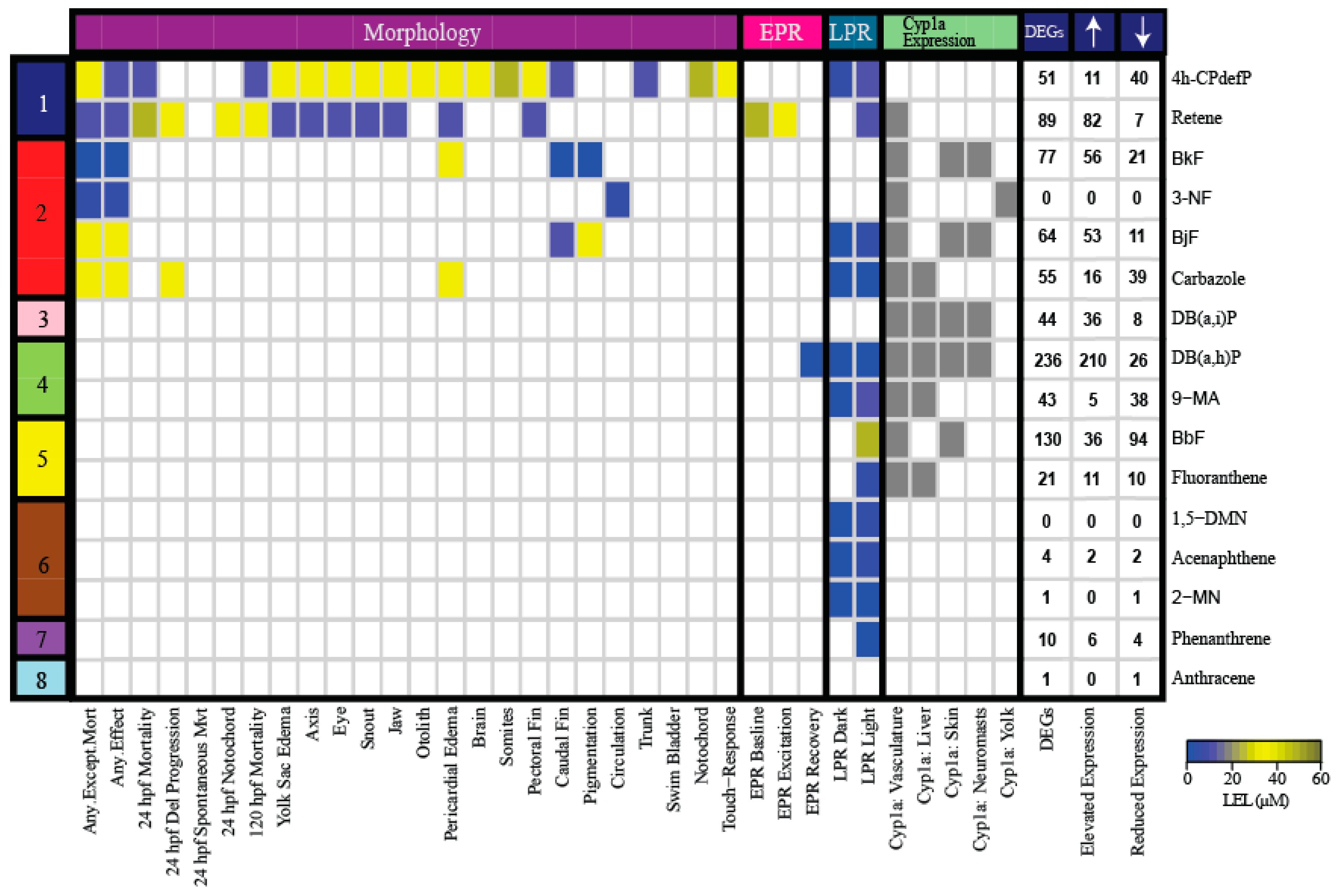
2.3. Number of Differentially Expressed Genes (DEGs) Generally Correlates with PAH Developmental Toxicity Profiles
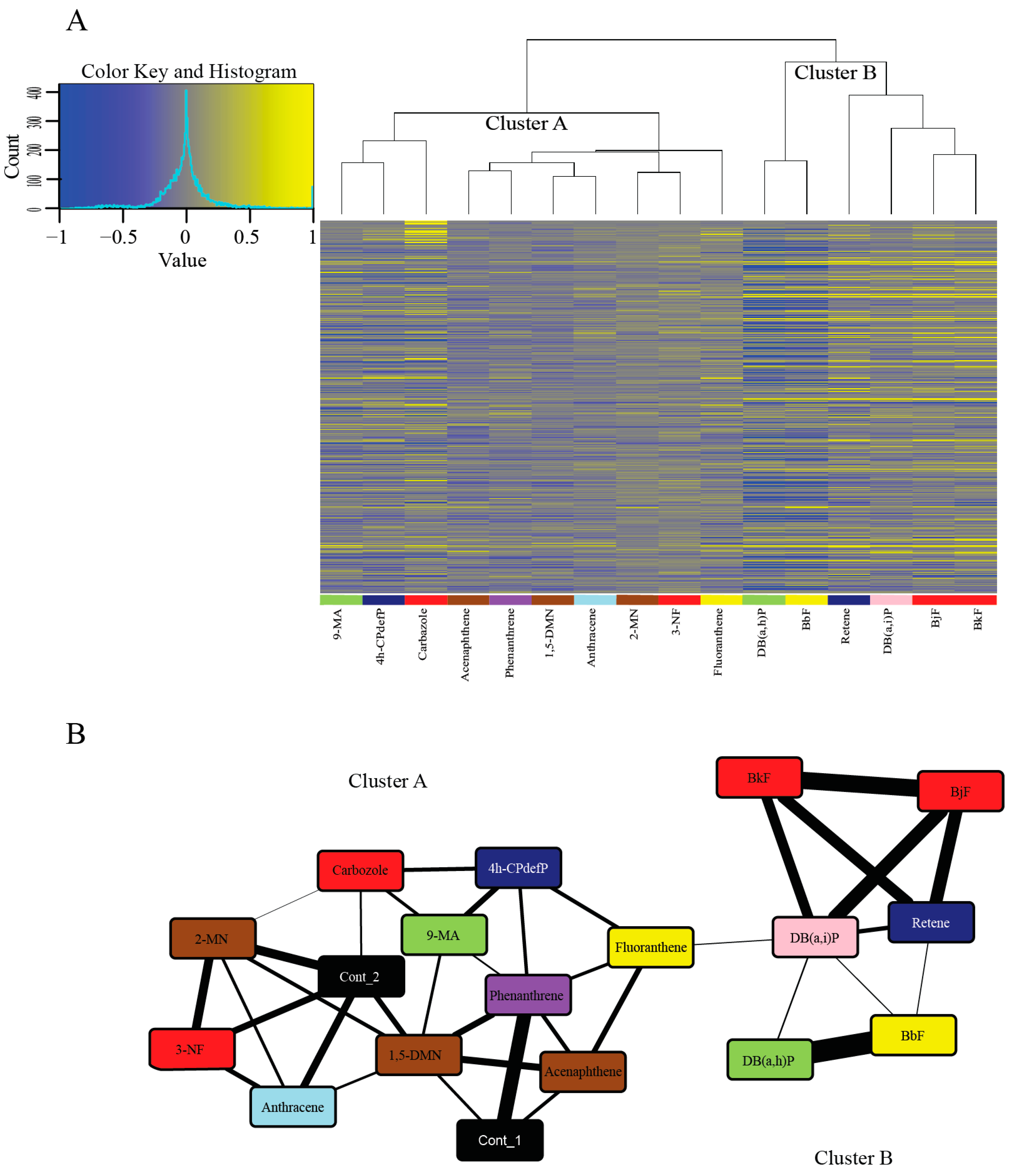
2.4. Cluster Analysis of Response to PAHs Reveals Two Broad Clusters
2.5. Cyp1a Expression is An Inadequate Biomarker for PAH Developmental Toxicity, but is Associated with Transcriptomic Response
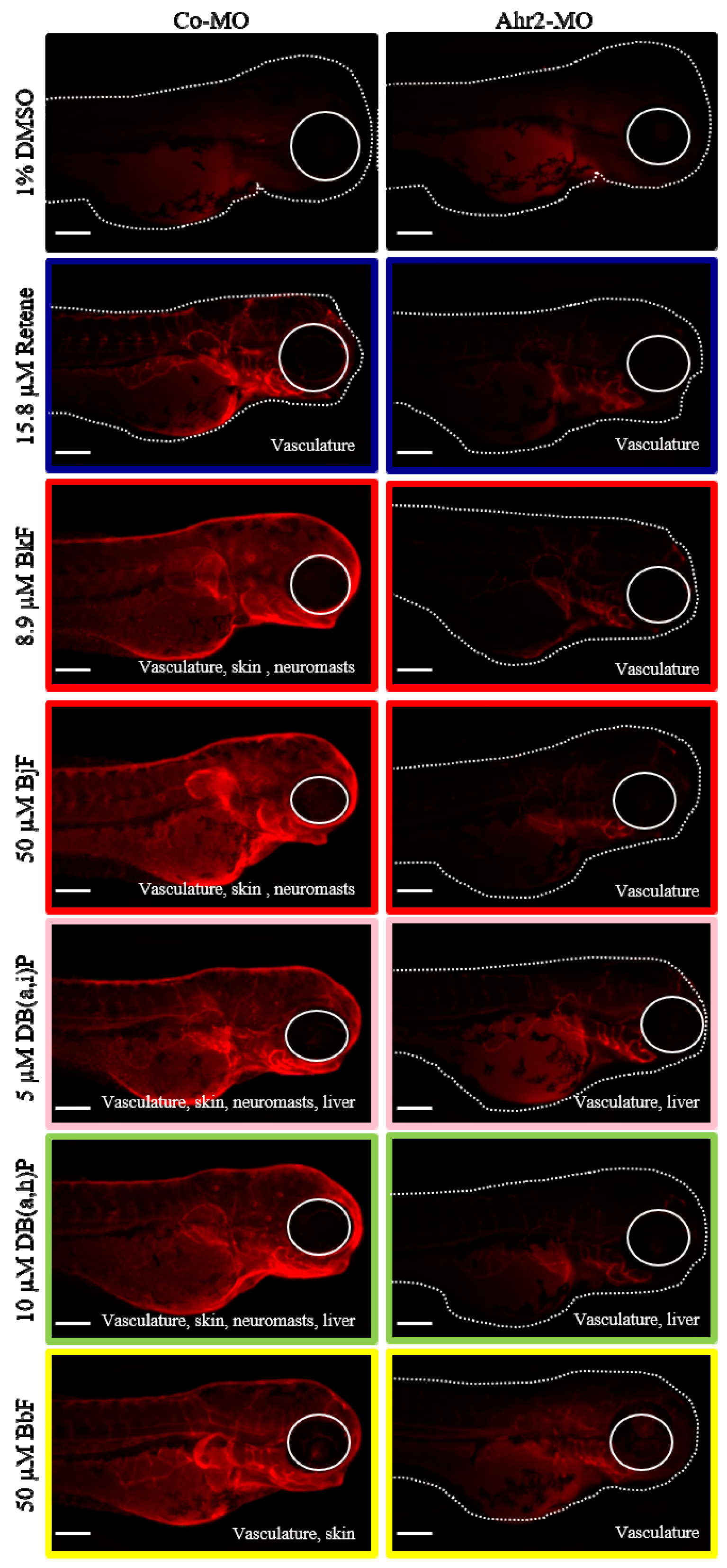
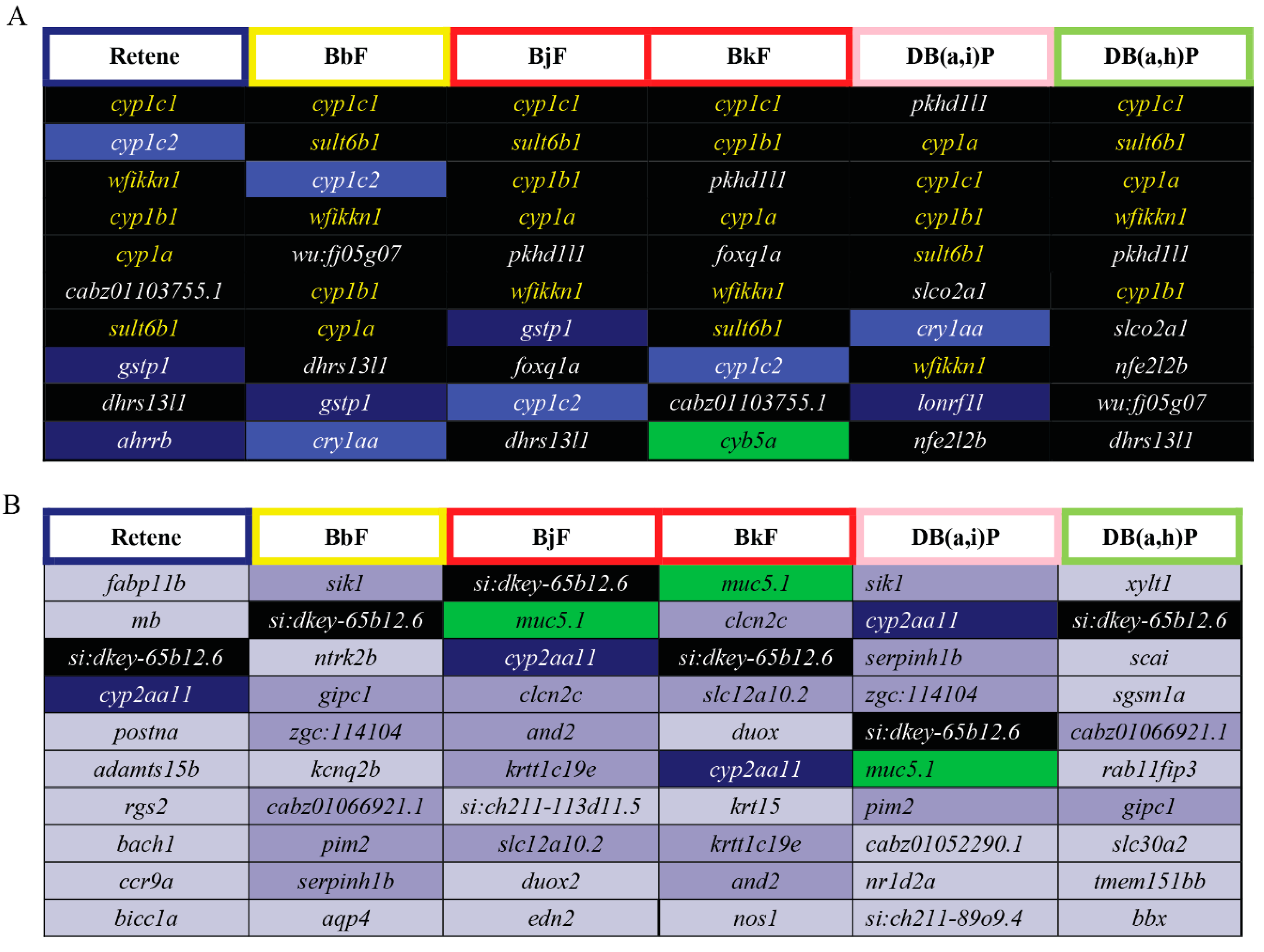
2.6. Cluster B PAHs Activated Ahr2
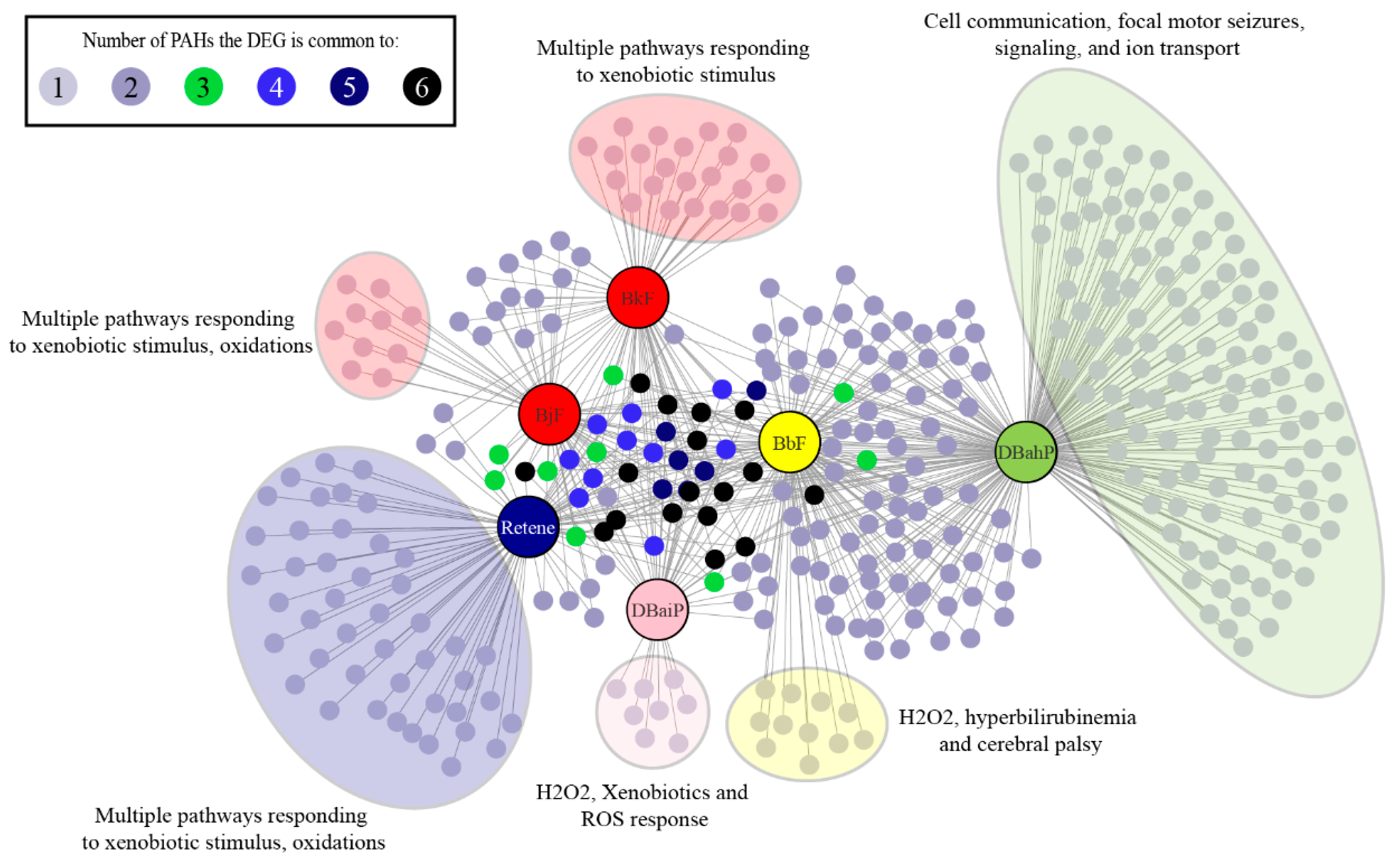
2.7. Cluster B PAHs that Activate Ahr2 Uniquely Enrich Several Pathways
2.8. Cluster B PAHs had Seventeen Common DEGs
3. Methods
3.1. Chemicals
3.1.1. Zebrafish Husbandry
3.1.2. Exposures
3.2. RNA Sequencing (RNA-seq)
3.2.1. Exposure Concentrations
3.2.2. Developmental Toxicity (Morphology) Assessments
3.2.3. RNA Isolation and Sequencing
3.2.4. Analysis of RNA-seq Data
3.2.5. Network Analysis
3.3. Immunohistochemistry
3.3.1. Morpholino Injections
3.3.2. Exposures and Immunohistochemistry (IHC)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, K.H.; Jahan, S.A.; Kabir, E.; Brown, R.J. A review of airborne polycyclic aromatic hydrocarbons (PAHs) and their human health effects. Environ. Int. 2013, 60, 71–80. [Google Scholar] [CrossRef]
- Abdel-Shafy, H.I.; Mansour, M.S. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egypt. J. Pet. 2016, 25, 107–123. [Google Scholar] [CrossRef]
- Liu, B.; Xue, Z.; Zhu, X.; Jia, C. Long-term trends (1990-2014), health risks, and sources of atmospheric polycyclic aromatic hydrocarbons (PAHs) in the U.S. Environ. Pollut. 2017, 220, 1171–1179. [Google Scholar] [CrossRef]
- Liu, H.; Yu, X.; Liu, Z.; Sun, Y. Occurrence, characteristics and sources of polycyclic aromatic hydrocarbons in arable soils of Beijing, China. Ecotoxicol. Environ. Saf. 2018, 159, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Sarria-Villa, R.; Ocampo-Duque, W.; Paez, M.; Schuhmacher, M. Presence of PAHs in water and sediments of the Colombian Cauca River during heavy rain episodes, and implications for risk assessment. Sci. Total Environ. 2016, 540, 455–465. [Google Scholar] [CrossRef]
- Hong, W.J.; Jia, H.; Li, Y.F.; Sun, Y.; Liu, X.; Wang, L. Polycyclic aromatic hydrocarbons (PAHs) and alkylated PAHs in the coastal seawater, surface sediment and oyster from Dalian, Northeast China. Ecotoxicol. Environ. Saf. 2016, 128, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kabziński, A.; Cyran, J.; Juszczak, R. Determination of polycyclic aromatic hydrocarbons in water (including drinking water) of Łódź. Pol. J. Environ. Stud. 2002, 11, 695–706. [Google Scholar]
- Desalme, D.; Binet, P.; Chiapusio, G. Challenges in tracing the fate and effects of atmospheric polycyclic aromatic hydrocarbon deposition in vascular plants. Environ. Sci. Technol. 2013, 47, 3967–3981. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Liu, W.; Tao, S. Emission of polycyclic aromatic hydrocarbons in China. Environ. Sci. Technol. 2006, 40, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.; Alam, M.S.; Harrison, R.M. Source apportionment of polycyclic aromatic hydrocarbons in urban air using positive matrix factorization and spatial distribution analysis. Atmos. Environ. 2013, 79, 271–285. [Google Scholar] [CrossRef]
- Nikolaou, K.; Masclet, P.; Mouvier, G. Sources and chemical reactivity of polynuclear aromatic hydrocarbons in the atmosphere—A critical review. Sci. Total. Environ. 1984, 32, 103–132. [Google Scholar] [CrossRef]
- Mastrangelo, G.; Fadda, E.; Marzia, V. Polycyclic aromatic hydrocarbons and cancer in man. Environ. Health Perspect 1996, 104, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, B.; Hutchinson, E.; Unwin, J.; Fletcher, T. Lung cancer risk after exposure to polycyclic aromatic hydrocarbons: A review and meta-analysis. Environ. Health Perspect 2004, 112, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.P.; Rauh, V.; Whyatt, R.M.; Tang, D.; Tsai, W.Y.; Bernert, J.T.; Tu, Y.H.; Andrews, H.; Barr, D.B.; Camann, D.E.; et al. A summary of recent findings on birth outcomes and developmental effects of prenatal ETS, PAH, and pesticide exposures. Neurotoxicology 2005, 26, 573–587. [Google Scholar] [CrossRef]
- Cavalieri, E.L.; Higginbotham, S.; RamaKrishna, N.V.; Devanesan, P.D.; Todorovic, R.; Rogan, E.G.; Salmasi, S. Comparative dose-response tumorigenicity studies of dibenzo[alpha,l]pyrene versus 7,12-dimethylbenz[alpha]anthracene, benzo[alpha]pyrene and two dibenzo[alpha,l]pyrene dihydrodiols in mouse skin and rat mammary gland. Carcinogenesis 1991, 12, 1939–1944. [Google Scholar] [CrossRef]
- Collins, J.F.; Brown, J.P.; Alexeeff, G.V.; Salmon, A.G. Potency equivalency factors for some polycyclic aromatic hydrocarbons and polycyclic aromatic hydrocarbon derivatives. Regul. Toxicol. Pharmacol. 1998, 28, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Norramit, P.; Cheevaporn, V.; Itoh, N.; Tanaka, K. Characterization and carcinogenic risk assessment of polycyclic aromatic hydrocarbons in the respirable fraction of airborne particles in the Bangkok Metropolitan area. J. Health Sci. 2005, 51, 437–446. [Google Scholar] [CrossRef][Green Version]
- Boffetta, P.; Jourenkova, N.; Gustavsson, P. Cancer risk from occupational and environmental exposure to polycyclic aromatic hydrocarbons. Cancer Cause Control 1997, 8, 444–472. [Google Scholar] [CrossRef]
- (ATSDR), A. f. T. S. a. D. R. Toxicological Profile for Polycyclic Aromatic Hydrocarbons (PAHs); Services, U.D. o. H. a. H., Ed.; Public Health Service: Atlanta, GA, USA, 1995. [Google Scholar]
- Samburova, V.; Zielinska, B.; Khlystov, A. Do 16 Polycyclic Aromatic Hydrocarbons Represent PAH Air Toxicity? Toxics 2017, 5, 17. [Google Scholar] [CrossRef]
- Andersson, J.T.; Achten, C. Time to say goodbye to the 16 EPA PAHs? Toward an up-to-date use of PACs for environmental purposes. Polycycl. Aromat. Compd. 2015, 35, 330–354. [Google Scholar] [CrossRef]
- Keith, L.H. The Source of US EPA’s Sixteen PAH Priority Pollutants. Polycycl. Aromat. Compd. 2015, 35, 147–160. [Google Scholar] [CrossRef]
- EPA, U. Development of a Relative Potency Factor (RPF) Approach for Polycyclic Aromatic Hydrocarbon (PAH) Mixtures. Intregrated Risk Inf. Syst. (IRIS) 2010. [Google Scholar]
- Schoeny, R.; Poirier, K. Provisional Guidance for Quantitative Risk Assessment of Polycyclic Aromatic Hydrocarbons; Final report; Environmental Protection Agency: Cincinnati, OH, USA, 1993. [Google Scholar]
- Perera, F.P.; Whyatt, R.M.; Jedrychowski, W.; Rauh, V.; Manchester, D.; Santella, R.M.; Ottman, R. Recent developments in molecular epidemiology: A study of the effects of environmental polycyclic aromatic hydrocarbons on birth outcomes in Poland. Am. J. Epidemiol. 1998, 147, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Perzanowski, M.; Rundle, A.; Moors, K.; Yan, B.; Chillrud, S.N.; Whyatt, R.; Camann, D.; Perera, F.P.; Miller, R.L. Polycyclic aromatic hydrocarbon exposure, obesity and childhood asthma in an urban cohort. Environ. Res. 2014, 128, 35–41. [Google Scholar] [CrossRef]
- Perera, F.P.; Rauh, V.; Whyatt, R.M.; Tsai, W.Y.; Tang, D.; Diaz, D.; Hoepner, L.; Barr, D.; Tu, Y.H.; Camann, D.; et al. Effect of prenatal exposure to airborne polycyclic aromatic hydrocarbons on neurodevelopment in the first 3 years of life among inner-city children. Environ. Health Perspect. 2006, 114, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Genkinger, J.M.; Stigter, L.; Jedrychowski, W.; Huang, T.J.; Wang, S.; Roen, E.L.; Majewska, R.; Kieltyka, A.; Mroz, E.; Perera, F.P. Prenatal polycyclic aromatic hydrocarbon (PAH) exposure, antioxidant levels and behavioral development of children ages 6-9. Environ. Res. 2015, 140, 136–144. [Google Scholar] [CrossRef]
- Jedrychowski, W.A.; Perera, F.P.; Camann, D.; Spengler, J.; Butscher, M.; Mroz, E.; Majewska, R.; Flak, E.; Jacek, R.; Sowa, A. Prenatal exposure to polycyclic aromatic hydrocarbons and cognitive dysfunction in children. Environ. Sci. Pollut. Res. Int. 2015, 22, 3631–3639. [Google Scholar] [CrossRef] [PubMed]
- Burstyn, I.; Kromhout, H.; Partanen, T.; Svane, O.; Langard, S.; Ahrens, W.; Kauppinen, T.; Stucker, I.; Shaham, J.; Heederik, D.; et al. Polycyclic aromatic hydrocarbons and fatal ischemic heart disease. Epidemiology 2005, 16, 744–750. [Google Scholar] [CrossRef]
- Burczynski, M.E.; Lin, H.K.; Penning, T.M. Isoform-specific induction of a human aldo-keto reductase by polycyclic aromatic hydrocarbons (PAHs), electrophiles, and oxidative stress: Implications for the alternative pathway of PAH activation catalyzed by human dihydrodiol dehydrogenase. Cancer Res. 1999, 59, 607–614. [Google Scholar] [PubMed]
- Billiard, S.M.; Timme-Laragy, A.R.; Wassenberg, D.M.; Cockman, C.; Di Giulio, R.T. The role of the aryl hydrocarbon receptor pathway in mediating synergistic developmental toxicity of polycyclic aromatic hydrocarbons to zebrafish. Toxicol. Sci. 2006, 92, 526–536. [Google Scholar] [CrossRef] [PubMed]
- Billiard, S.M.; Meyer, J.N.; Wassenberg, D.M.; Hodson, P.V.; Di Giulio, R.T. Nonadditive effects of PAHs on Early Vertebrate Development: Mechanisms and implications for risk assessment. Toxicol. Sci. 2008, 105, 5–23. [Google Scholar] [CrossRef]
- Knecht, A.L.; Goodale, B.C.; Truong, L.; Simonich, M.T.; Swanson, A.J.; Matzke, M.M.; Anderson, K.A.; Waters, K.M.; Tanguay, R.L. Comparative developmental toxicity of environmentally relevant oxygenated PAHs. Toxicol. Appl. Pharmacol. 2013, 271, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Jayasundara, N.; Van Tiem Garner, L.; Meyer, J.N.; Erwin, K.N.; Di Giulio, R.T. AHR2-Mediated transcriptomic responses underlying the synergistic cardiac developmental toxicity of PAHs. Toxicol. Sci. 2015, 143, 469–481. [Google Scholar] [CrossRef]
- Xu, X.; Cook, R.L.; Ilacqua, V.A.; Kan, H.; Talbott, E.O.; Kearney, G. Studying associations between urinary metabolites of polycyclic aromatic hydrocarbons (PAHs) and cardiovascular diseases in the United States. Sci. Total. Environ. 2010, 408, 4943–4948. [Google Scholar] [CrossRef]
- Qiao, M.; Qi, W.; Liu, H.; Qu, J. Simultaneous determination of typical substituted and parent polycyclic aromatic hydrocarbons in water and solid matrix by gas chromatography-mass spectrometry. J. Chromatogr. A 2013, 1291, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Tanguay, R.L.; Abnet, C.C.; Heideman, W.; Peterson, R.E. Cloning and characterization of the zebrafish (Danio rerio) aryl hydrocarbon receptor. Biochim. Biophys. Acta. 1999, 1444, 35–48. [Google Scholar] [CrossRef]
- Prasch, A.L.; Tanguay, R.L.; Mehta, V.; Heideman, W.; Peterson, R.E. Identification of zebrafish ARNT1 homologs: 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in the developing zebrafish requires ARNT1. Mol. Pharmacol. 2006, 69, 776–787. [Google Scholar]
- Yoshioka, W.; Peterson, R.E.; Tohyama, C. Molecular targets that link dioxin exposure to toxicity phenotypes. J. Steroid Biochem. Mol. Biol. 2011, 127, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.; Harper, S.L.; Tanguay, R.L. Evaluation of embryotoxicity using the zebrafish model. Methods Mol. Biol. 2011, 691, 271–279. [Google Scholar]
- Udvadia, A.J.; Linney, E. Windows into development: Historic, current, and future perspectives on transgenic zebrafish. Dev. Biol. 2003, 256, 1–17. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Collier, T.K.; Scholz, N.L. Defects in cardiac function precede morphological abnormalities in fish embryos exposed to polycyclic aromatic hydrocarbons. Toxicol. Appl. Pharmacol. 2004, 196, 191–205. [Google Scholar] [CrossRef]
- Geier, M.C.; Chlebowski, A.C.; Truong, L.; Massey Simonich, S.L.; Anderson, K.A.; Tanguay, R.L. Comparative developmental toxicity of a comprehensive suite of polycyclic aromatic hydrocarbons. Arch. Toxicol. 2018, 92, 571–586. [Google Scholar] [CrossRef]
- Knecht, A.L.; Truong, L.; Simonich, M.T.; Tanguay, R.L. Developmental benzo[a]pyrene (B[a]P) exposure impacts larval behavior and impairs adult learning in zebrafish. Neurotoxicol. Teratol. 2017, 59, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Chlebowski, A.C.; Garcia, G.R.; La Du, J.K.; Bisson, W.H.; Truong, L.; Massey Simonich, S.L.; Tanguay, R.L. Mechanistic Investigations Into the Developmental Toxicity of Nitrated and Heterocyclic PAHs. Toxicol. Sci. 2017, 157, 246–259. [Google Scholar] [CrossRef]
- Brinkmann, M.; Koglin, S.; Eisner, B.; Wiseman, S.; Hecker, M.; Eichbaum, K.; Thalmann, B.; Buchinger, S.; Reifferscheid, G.; Hollert, H. Characterisation of transcriptional responses to dioxins and dioxin-like contaminants in roach (Rutilus rutilus) using whole transcriptome analysis. Sci. Total Environ. 2016, 541, 412–423. [Google Scholar] [CrossRef]
- Goodale, B.C.; La Du, J.; Tilton, S.C.; Sullivan, C.M.; Bisson, W.H.; Waters, K.M.; Tanguay, R.L. Ligand-Specific Transcriptional Mechanisms Underlie Aryl Hydrocarbon Receptor-Mediated Developmental Toxicity of Oxygenated PAHs. Toxicol. Sci. 2015, 147, 397–411. [Google Scholar] [CrossRef]
- Goodale, B.C.; Tilton, S.C.; Corvi, M.M.; Wilson, G.R.; Janszen, D.B.; Anderson, K.A.; Waters, K.M.; Tanguay, R.L. Structurally distinct polycyclic aromatic hydrocarbons induce differential transcriptional responses in developing zebrafish. Toxicol. Appl. Pharmacol. 2013, 272, 656–670. [Google Scholar] [CrossRef]
- Nebert, D.W.; Dalton, T.P.; Okey, A.B.; Gonzalez, F.J. Role of aryl hydrocarbon receptor-mediated induction of the CYP1 enzymes in environmental toxicity and cancer. J. Biol. Chem. 2004, 279, 23847–23850. [Google Scholar] [CrossRef]
- Andreasen, E.A.; Hahn, M.E.; Heideman, W.; Peterson, R.E.; Tanguay, R.L. The zebrafish (Danio rerio) aryl hydrocarbon receptor type 1 is a novel vertebrate receptor. Mol. Pharmacol. 2002, 62, 234–249. [Google Scholar] [CrossRef]
- Karchner, S.I.; Franks, D.G.; Hahn, M.E. AHR1B, a new functional aryl hydrocarbon receptor in zebrafish: Tandem arrangement of ahr1b and ahr2 genes. Biochem. J. 2005. [Google Scholar] [CrossRef]
- Goodale, B.C.; La Du, J.K.; Bisson, W.H.; Janszen, D.B.; Waters, K.M.; Tanguay, R.L. AHR2 mutant reveals functional diversity of aryl hydrocarbon receptors in zebrafish. PLoS ONE 2012, 7, e29346. [Google Scholar] [CrossRef]
- Incardona, J.P.; Day, H.L.; Collier, T.K.; Scholz, N.L. Developmental toxicity of 4-ring polycyclic aromatic hydrocarbons in zebrafish is differentially dependent on AH receptor isoforms and hepatic cytochrome P4501A metabolism. Toxicol. Appl. Pharmacol. 2006, 217, 308–321. [Google Scholar] [CrossRef]
- Incardona, J.P.; Carls, M.G.; Teraoka, H.; Sloan, C.A.; Collier, T.K.; Scholz, N.L. Aryl hydrocarbon receptor-independent toxicity of weathered crude oil during fish development. Environ. Health Perspect. 2005, 113, 1755–1762. [Google Scholar] [CrossRef]
- Wang, W.; Jariyasopit, N.; Schrlau, J.; Jia, Y.; Tao, S.; Yu, T.W.; Dashwood, R.H.; Zhang, W.; Wang, X.; Simonich, S.L. Concentration and photochemistry of PAHs, NPAHs, and OPAHs and toxicity of PM2.5 during the Beijing Olympic Games. Environ. Sci. Technol. 2011, 45, 6887–6895. [Google Scholar] [CrossRef]
- Wincent, E.; Jonsson, M.E.; Bottai, M.; Lundstedt, S.; Dreij, K. Aryl hydrocarbon receptor activation and developmental toxicity in zebrafish in response to soil extracts containing unsubstituted and oxygenated PAHs. Environ. Sci. Technol. 2015, 49, 3869–3877. [Google Scholar] [CrossRef]
- Brinkmann, M.; Maletz, S.; Krauss, M.; Bluhm, K.; Schiwy, S.; Kuckelkorn, J.; Tiehm, A.; Brack, W.; Hollert, H. Heterocyclic aromatic hydrocarbons show estrogenic activity upon metabolization in a recombinant transactivation assay. Environ. Sci. Technol. 2014, 48, 5892–5901. [Google Scholar] [CrossRef]
- Chlebowski, A.C.; Tanguay, R.L.; Simonich, S.L. Quantitation and prediction of sorptive losses during toxicity testing of polycyclic aromatic hydrocarbon (PAH) and nitrated PAH (NPAH) using polystyrene 96-well plates. Neurotoxicol. Teratol. 2016, 57, 30–38. [Google Scholar] [CrossRef][Green Version]
- Schreiber, R.; Altenburger, R.; Paschke, A.; Kuster, E. How to deal with lipophilic and volatile organic substances in microtiter plate assays. Environ. Toxicol. Chem. 2008, 27, 1676–1682. [Google Scholar] [CrossRef]
- Wolska, L.; Rawa-Adkonis, M.; Namiesnik, J. Determining PAHs and PCBs in aqueous samples: Finding and evaluating sources of error. Anal. Bioanal. Chem. 2005, 382, 1389–1397. [Google Scholar] [CrossRef]
- Minick, D.J.; Anderson, K.A. Diffusive flux of PAHs across sediment-water and water-air interfaces at urban superfund sites. Environ. Toxicol. Chem. 2017, 36, 2281–2289. [Google Scholar] [CrossRef]
- Tidwell, L.G.; Blair Paulik, L.; Anderson, K.A. Air-water exchange of PAHs and OPAHs at a superfund mega-site. Sci. Total. Environ. 2017, 603–604, 676–686. [Google Scholar] [CrossRef]
- Truong, L.; Reif, D.M.; St Mary, L.; Geier, M.C.; Truong, H.D.; Tanguay, R.L. Multidimensional in vivo hazard assessment using zebrafish. Toxicol. Sci. 2014, 137, 212–233. [Google Scholar] [CrossRef]
- Haggard, D.E.; Das, S.R.; Tanguay, R.L. Comparative Toxicogenomic Responses to the Flame Retardant mITP in Developing Zebrafish. Chem. Res. Toxicol. 2017, 30, 508–515. [Google Scholar] [CrossRef]
- Hawliczek, A.; Nota, B.; Cenijn, P.; Kamstra, J.; Pieterse, B.; Winter, R.; Winkens, K.; Hollert, H.; Segner, H.; Legler, J. Developmental toxicity and endocrine disrupting potency of 4-azapyrene, benzo[b]fluorene and retene in the zebrafish Danio rerio. Reprod. Toxicol. 2012, 33, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Geier, M.C.; James Minick, D.; Truong, L.; Tilton, S.; Pande, P.; Anderson, K.A.; Teeguardan, J.; Tanguay, R.L. Systematic developmental neurotoxicity assessment of a representative PAH Superfund mixture using zebrafish. Toxicol. Appl. Pharmacol. 2018, 354, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Achten, C.; Andersson, J.T. Overview of Polycyclic Aromatic Compounds (PAC). Polycycl. Aromat. Compd. 2015, 35, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Du, Y.; Lam, P.K.; Wu, R.S.; Zhou, B. Developmental toxicity and alteration of gene expression in zebrafish embryos exposed to PFOS. Toxicol. Appl. Pharmacol. 2008, 230, 23–32. [Google Scholar] [CrossRef]
- Reimers, M.J.; Flockton, A.R.; Tanguay, R.L. Ethanol- and acetaldehyde-mediated developmental toxicity in zebrafish. Neurotoxicol. Teratol. 2004, 26, 769–781. [Google Scholar] [CrossRef]
- Ober, E.A.; Field, H.A.; Stainier, D.Y. From endoderm formation to liver and pancreas development in zebrafish. Mech. Dev. 2003, 120, 5–18. [Google Scholar] [CrossRef]
- Van der Oost, R.; Beyer, J.; Vermeulen, N.P. Fish bioaccumulation and biomarkers in environmental risk assessment: A review. Environ. Toxicol. Pharmacol. 2003, 13, 57–149. [Google Scholar] [CrossRef]
- Lundstedt, S.; White, P.A.; Lemieux, C.L.; Lynes, K.D.; Lambert, I.B.; Oberg, L.; Haglund, P.; Tysklind, M. Sources, fate, and toxic hazards of oxygenated polycyclic aromatic hydrocarbons (PAHs) at PAH-contaminated sites. Ambio 2007, 36, 475–485. [Google Scholar] [CrossRef]
- Goldstone, J.V.; McArthur, A.G.; Kubota, A.; Zanette, J.; Parente, T.; Jonsson, M.E.; Nelson, D.R.; Stegeman, J.J. Identification and developmental expression of the full complement of Cytochrome P450 genes in Zebrafish. BMC Genom. 2010, 11, 643. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-J.; Hsiao, P.; Kuo, M.-W.; Chung, B.-C. Expression of zebrafish cyp11a1 as a maternal transcript and in yolk syncytial layer. Gene Expr. Patterns 2002, 2, 219–222. [Google Scholar] [CrossRef]
- Incardona, J.P.; Linbo, T.L.; Scholz, N.L. Cardiac toxicity of 5-ring polycyclic aromatic hydrocarbons is differentially dependent on the aryl hydrocarbon receptor 2 isoform during zebrafish development. Toxicol. Appl. Pharmacol. 2011, 257, 242–249. [Google Scholar] [CrossRef]
- Garcia, G.R.; Bugel, S.M.; Truong, L.; Spagnoli, S.; Tanguay, R.L. AHR2 required for normal behavioral responses and proper development of the skeletal and reproductive systems in zebrafish. PLoS ONE 2018, 13, e0193484. [Google Scholar] [CrossRef]
- Kubota, A.; Goldstone, J.V.; Lemaire, B.; Takata, M.; Woodin, B.R.; Stegeman, J.J. Pregnane X receptor and aryl hydrocarbon receptor both are involved in transcriptional regulation of pxr, CYP2 and CYP3 genes in developing zebrafish. Toxicol. Sci. 2014. [Google Scholar]
- Prasch, A.L.; Teraoka, H.; Carney, S.A.; Dong, W.; Hiraga, T.; Stegeman, J.J.; Heideman, W.; Peterson, R.E. Aryl hydrocarbon receptor 2 mediates 2,3,7,8-tetrachlorodibenzo-p-dioxin developmental toxicity in zebrafish. Toxicol. Sci. 2003, 76, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Bello, S.M.; Prasch, A.L.; Peterson, R.E.; Heideman, W. Water permeability and TCDD-induced edema in zebrafish early-life stages. Toxicol. Sci. 2004, 78, 78–87. [Google Scholar] [CrossRef]
- Scott, J.A.; Incardona, J.P.; Pelkki, K.; Shepardson, S.; Hodson, P.V. AhR2-mediated, CYP1A-independent cardiovascular toxicity in zebrafish (Danio rerio) embryos exposed to retene. Aquat. Toxicol. 2011, 101, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Geier, M.C.; Garland, M.A.; Bugel, S.M.; Dunham, C.L.; Brown, J.; Tilton, S.C.; Tanguay, R.L. Benzofluoranthenes produce ectopic fin development in zebrafish revealing the complexity of AHR-dependent PAH activity. Manuscript in prepration.
- Reimand, J.; Arak, T.; Adler, P.; Kolberg, L.; Reisberg, S.; Peterson, H.; Vilo, J. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016, 44, W83–W89. [Google Scholar] [CrossRef]
- Wang, Q.; Liang, K.; Liu, J.; Yang, L.; Guo, Y.; Liu, C.; Zhou, B. Exposure of zebrafish embryos/larvae to TDCPP alters concentrations of thyroid hormones and transcriptions of genes involved in the hypothalamic-pituitary-thyroid axis. Aquat. Toxicol. 2013, 126, 207–213. [Google Scholar] [CrossRef]
- Zhai, W.; Huang, Z.; Chen, L.; Feng, C.; Li, B.; Li, T. Thyroid endocrine disruption in zebrafish larvae after exposure to mono-(2-ethylhexyl) phthalate (MEHP). PLoS ONE 2014, 9, e92465. [Google Scholar] [CrossRef]
- Huang, G.M.; Tian, X.F.; Fang, X.D.; Ji, F.J. Waterborne exposure to bisphenol F causes thyroid endocrine disruption in zebrafish larvae. Chemosphere 2016, 147, 188–194. [Google Scholar] [CrossRef]
- Takahashi, S.; Sakakibara, Y.; Mishiro, E.; Kouriki, H.; Nobe, R.; Kurogi, K.; Yasuda, S.; Liu, M.C.; Suiko, M. Molecular cloning, expression and characterization of a novel mouse SULT6 cytosolic sulfotransferase. J. Biochem. 2009, 146, 399–405. [Google Scholar] [CrossRef]
- Bui, A.; Xiao, R.; Perveen, Z.; Kleinow, K.; Penn, A. Zebrafish embryos sequester and retain petrochemical combustion products: Developmental and transcriptome consequences. Aquat Toxicol 2012, 108, 23–32. [Google Scholar] [CrossRef]
- Maikawa, C.L.; Zimmerman, N.; Rais, K.; Shah, M.; Hawley, B.; Pant, P.; Jeong, C.H.; Delgado-Saborit, J.M.; Volckens, J.; Evans, G.; et al. Murine precision-cut lung slices exhibit acute responses following exposure to gasoline direct injection engine emissions. Sci. Total. Environ. 2016, 568, 1102–1109. [Google Scholar] [CrossRef]
- Dalton, T.P.; Puga, A.; Shertzer, H.G. Induction of cellular oxidative stress by aryl hydrocarbon receptor activation. Chem. Biol. Interact. 2002, 141, 77–95. [Google Scholar] [CrossRef]
- Duan, J.; Yu, Y.; Li, Y.; Jing, L.; Yang, M.; Wang, J.; Li, Y.; Zhou, X.; Miller, M.R.; Sun, Z. Comprehensive understanding of PM2.5 on gene and microRNA expression patterns in zebrafish (Danio rerio) model. Sci. Total Environ. 2017, 586, 666–674. [Google Scholar] [CrossRef]
- Xu, H.; Lam, S.H.; Shen, Y.; Gong, Z. Genome-wide identification of molecular pathways and biomarkers in response to arsenic exposure in zebrafish liver. PLoS ONE 2013, 8, e68737. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, C.; Wang, C.; Zhou, Y.; Gao, D.; Zuo, Z. Maternal and embryonic exposure to the water soluble fraction of crude oil or lead induces behavioral abnormalities in zebrafish (Danio rerio), and the mechanisms involved. Chemosphere 2018, 191, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Legradi, J.; El Abdellaoui, N.; van Pomeren, M.; Legler, J. Comparability of behavioural assays using zebrafish larvae to assess neurotoxicity. Environ. Sci. Pollut. Res. Int. 2015, 22, 16277–16289. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.; Cavanaugh, K.; Verbueken, E.; Pype, C.; Casteleyn, C.; Van Ginneken, C.; Van Cruchten, S. Xenobiotic metabolism in the zebrafish: A review of the spatiotemporal distribution, modulation and activity of Cytochrome P450 families 1 to 3. J. Toxicol. Sci. 2016, 41, 1–11. [Google Scholar] [CrossRef]
- Barton, C.L.; Johnson, E.W.; Tanguay, R.L. Facility Design and Health Management Program at the Sinnhuber Aquatic Research Laboratory. Zebrafish 2016, 13, S39–S43. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish. Available online: http://zfin.org/zf_info/zfbook/zfbk.html (accessed on 25 May 2019).
- Mandrell, D.; Truong, L.; Jephson, C.; Sarker, M.R.; Moore, A.; Lang, C.; Simonich, M.T.; Tanguay, R.L. Automated zebrafish chorion removal and single embryo placement: Optimizing throughput of zebrafish developmental toxicity screens. J. Lab. Autom. 2012, 17, 66–74. [Google Scholar] [CrossRef]
- Truong, L.; Bugel, S.M.; Chlebowski, A.; Usenko, C.Y.; Simonich, M.T.; Simonich, S.L.; Tanguay, R.L. Optimizing multi-dimensional high throughput screening using zebrafish. Reprod. Toxicol. 2016, 65, 139–147. [Google Scholar] [CrossRef]
- Kokel, D.; Dunn, T.W.; Ahrens, M.B.; Alshut, R.; Cheung, C.Y.; Saint-Amant, L.; Bruni, G.; Mateus, R.; van Ham, T.J.; Shiraki, T.; et al. Identification of nonvisual photomotor response cells in the vertebrate hindbrain. J. Neurosci. 2013, 33, 3834–3843. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; Version 3.4.1; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic Acids Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Faith, J.J.; Hayete, B.; Thaden, J.T.; Mogno, I.; Wierzbowski, J.; Cottarel, G.; Kasif, S.; Collins, J.J.; Gardner, T.S. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007, 5, e8. [Google Scholar] [CrossRef]
- Meyer, P.E.; Lafitte, F.; Bontempi, G. minet: A R/Bioconductor package for inferring large transcriptional networks using mutual information. BMC Bioinform. 2008, 9, 461. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.P.; Hseu, T.H.; Buhler, D.R.; Wang, W.D.; Hu, C.H. Constitutive and xenobiotics-induced expression of a novel CYP3A gene from zebrafish larva. Toxicol. Appl. Pharmacol. 2005, 20, 247–258. [Google Scholar] [CrossRef]
- Mathew, L.K.; Andreasen, E.A.; Tanguay, R.L. Aryl hydrocarbon receptor activation inhibits regenerative growth. Mol. Pharmacol. 2006, 69, 257–265. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bin # | Endpoints | ||
|---|---|---|---|
| Morphology | Behavior | Cyp1a | |
| 1 | x | x | x |
| 2 | x | x | x |
| 3 | x | x | |
| 4 | x | x | |
| 5 | x | x | |
| 6 | x | ||
| 7 | x | ||
| 8 | |||
| PAH with Abbreviation | Structure | CAS | Supplier | Purity (%) | Nominal Stock Conc. (mM) | RNA-seq Conc. (µM) | IHC Conc. (µM) |
|---|---|---|---|---|---|---|---|
| 4H-cyclopenta[def]phenanthren-4-one (4h-CPdefP) |  | 5737-13-3 | Chiron | 99.4 | 10 | 16.2 | 0.4 |
| Retene |  | 483-65-8 | SCB | 98 | 10 | 12.2 | 15.8 |
| Benzo[k]fluoranthene (BkF) |  | 207-08-9 | AccuStandard | 100 | 10 | 1.9 | 8.9 |
| 3-nitrofluoranthene (3-NF) |  | 892-21-7 | AccuStandard | 100 | 10 | 1.9 | 50 |
| Benzo[j]fluoranthene (BjF) |  | 205-82-3 | AccuStandard | 98.1 | 10 | 14.9 | 50 |
| Carbazole |  | 86-74-8 | AccuStandard | 99.7 | 10 | 50 | 35.6 |
| Dibenzo[a,i]pyrene (DB(a,i)P) |  | 189-55-9 | AccuStandard | 99.8 | 1 | 5 | 5 |
| Dibenzo[a,h]pyrene (DB(a,h)P) |  | 189-64-0 | AccuStandard | 99.9 | 10 | 10 | 10 |
| 9-methylanthracene (9-MA) |  | 779-02-2 | AccuStandard | 100 | 10 | 50 | 50 |
| Benzo[b]fluoranthene (BbF) |  | 205-99-2 | AccuStandard | 99.2 | 10 | 50 | 50 |
| Fluoranthene |  | 206-44-0 | AccuStandard | 97.2 | 10 | 50 | 50 |
| 1,5-dimethylnaphthalene (1,5-DMN) |  | 571-61-9 | AccuStandard | 100 | 10 | 50 | 50 |
| Acenaphthene |  | 83-32-9 | AccuStandard | 100 | 10 | 50 | 50 |
| 2-methylnaphthalene (2-MN) |  | 91-57-6 | AccuStandard | 100 | 10 | 50 | 50 |
| Phenanthrene |  | 65996-93-2 | AccuStandard | 99.5 | 10 | 50 | 50 |
| Anthracene |  | 120-12-7 | AccuStandard | 99.6 | 10 | 50 | 50 |
| Bin | PAH | Cyp1a log2FC | PADJ |
|---|---|---|---|
| Cluster A | |||
| 1 | 4h-CPdefP | 0.09 | 0.76 |
| 2 | 3-NF | 0.24 | 0.82 |
| Carbazole | 0.15 | 0.62 | |
| 4 | 9-MA | 0.31 | 0.13 |
| 5 | Fluoranthene | 0.26 | 0.34 |
| 6 | 1,5-DMN | 0.04 | 0.76 |
| Acenaphthene | 0.16 | 0.67 | |
| 2-MN | 0.02 | 0.99 | |
| 7 | Phenanthrene | 0.11 | 0.86 |
| 8 | Anthracene | 0.33 | 0.99 |
| Cluster B | |||
| 1 | Retene | 2.06 | 3.05E-57 |
| 2 | BkF | 2.18 | 2.00E-64 |
| BjF | 2.08 | 1.44E-58 | |
| 3 | DB(a,i)P | 1.37 | 1.84E-24 |
| 4 | DB(a,h)P | 1.22 | 1.09E-19 |
| 5 | BbF | 1.16 | 2.71E-17 |
| PAH Abbreviation | Concentrations (µM) Tested to Compute the EC80 |
|---|---|
| 4h-CPdefP | 8.5, 11.2, 13.9, 16.7, 19.4, 22.1, 24.8, 27.5, 30.2, 32.9, 35.6 |
| Retene | 2, 4, 6, 8, 10, 12, 14, 16, 18, 20, 22 |
| BkF | 0.5, 1.5, 2.5, 3.5, 4.5, 5.5, 6.5, 7.5, 8.5, 9.5, 10.5 |
| 3-NF | 0.25, 1, 2.1, 3.3, 4.4, 5.5, 6.6, 7.8, 8.9, 10.1, 11.2 |
| BjF | 4.5, 6.9, 9.3, 11.7, 14.1, 16.5, 18.8, 21.2, 23.6, 26, 28.4 |
| Carbazole | 8.5, 12.7, 16.8, 21, 25.1, 29.3, 33.4, 37.6, 41.7, 45.9, 50 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shankar, P.; Geier, M.C.; Truong, L.; McClure, R.S.; Pande, P.; Waters, K.M.; Tanguay, R.L. Coupling Genome-wide Transcriptomics and Developmental Toxicity Profiles in Zebrafish to Characterize Polycyclic Aromatic Hydrocarbon (PAH) Hazard. Int. J. Mol. Sci. 2019, 20, 2570. https://doi.org/10.3390/ijms20102570
Shankar P, Geier MC, Truong L, McClure RS, Pande P, Waters KM, Tanguay RL. Coupling Genome-wide Transcriptomics and Developmental Toxicity Profiles in Zebrafish to Characterize Polycyclic Aromatic Hydrocarbon (PAH) Hazard. International Journal of Molecular Sciences. 2019; 20(10):2570. https://doi.org/10.3390/ijms20102570
Chicago/Turabian StyleShankar, Prarthana, Mitra C. Geier, Lisa Truong, Ryan S. McClure, Paritosh Pande, Katrina M. Waters, and Robert L. Tanguay. 2019. "Coupling Genome-wide Transcriptomics and Developmental Toxicity Profiles in Zebrafish to Characterize Polycyclic Aromatic Hydrocarbon (PAH) Hazard" International Journal of Molecular Sciences 20, no. 10: 2570. https://doi.org/10.3390/ijms20102570
APA StyleShankar, P., Geier, M. C., Truong, L., McClure, R. S., Pande, P., Waters, K. M., & Tanguay, R. L. (2019). Coupling Genome-wide Transcriptomics and Developmental Toxicity Profiles in Zebrafish to Characterize Polycyclic Aromatic Hydrocarbon (PAH) Hazard. International Journal of Molecular Sciences, 20(10), 2570. https://doi.org/10.3390/ijms20102570