Molecular Characterization of Influenza Strains in Patients Admitted to Intensive Care Units during the 2017–2018 Season

 , , ,
, , , 
Abstract
:1. Introduction
2. Results
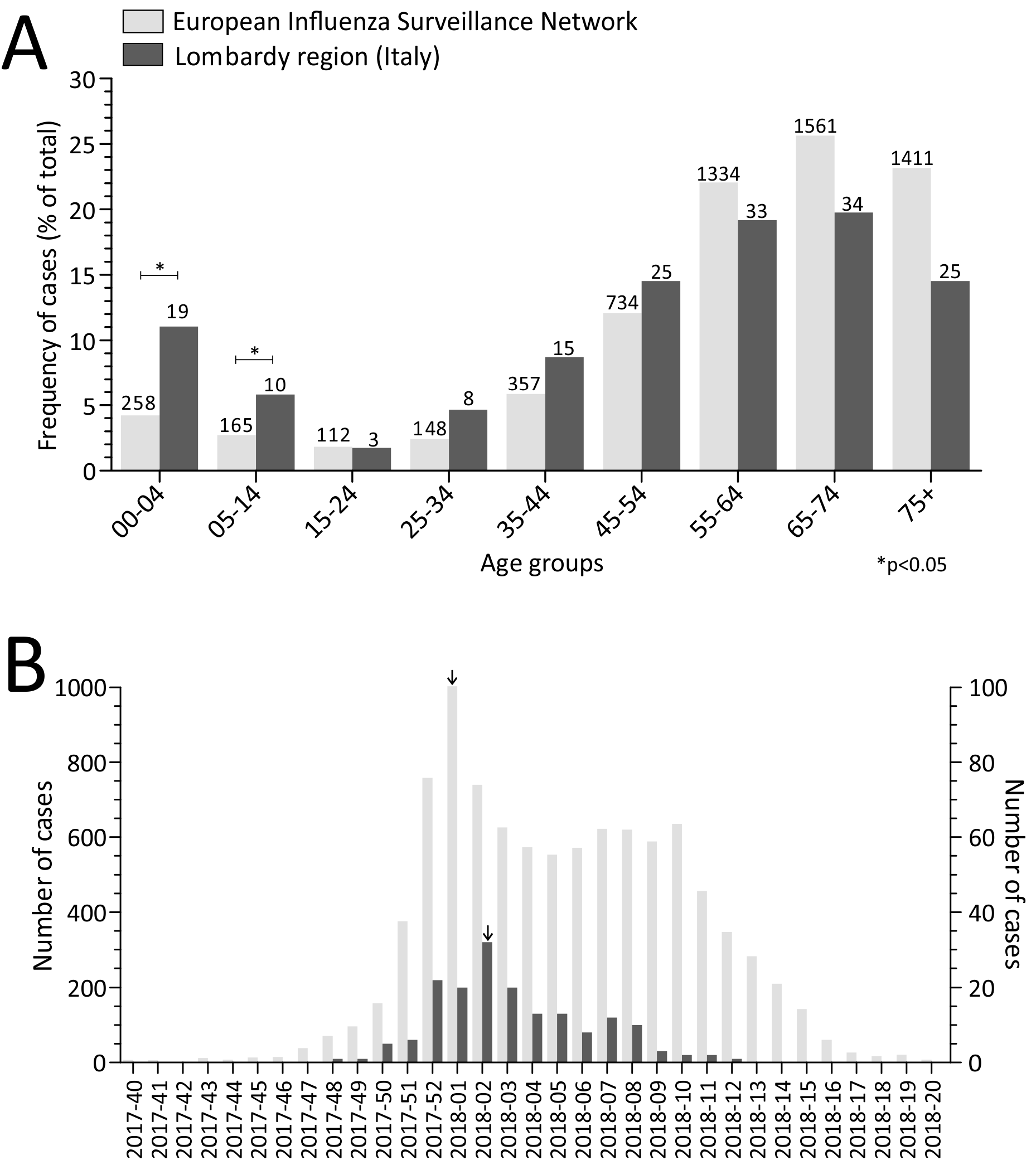
2.1. Laboratory-Confirmed Influenza ICU Cases
2.2. Temporal Distribution of Severe Influenza Cases
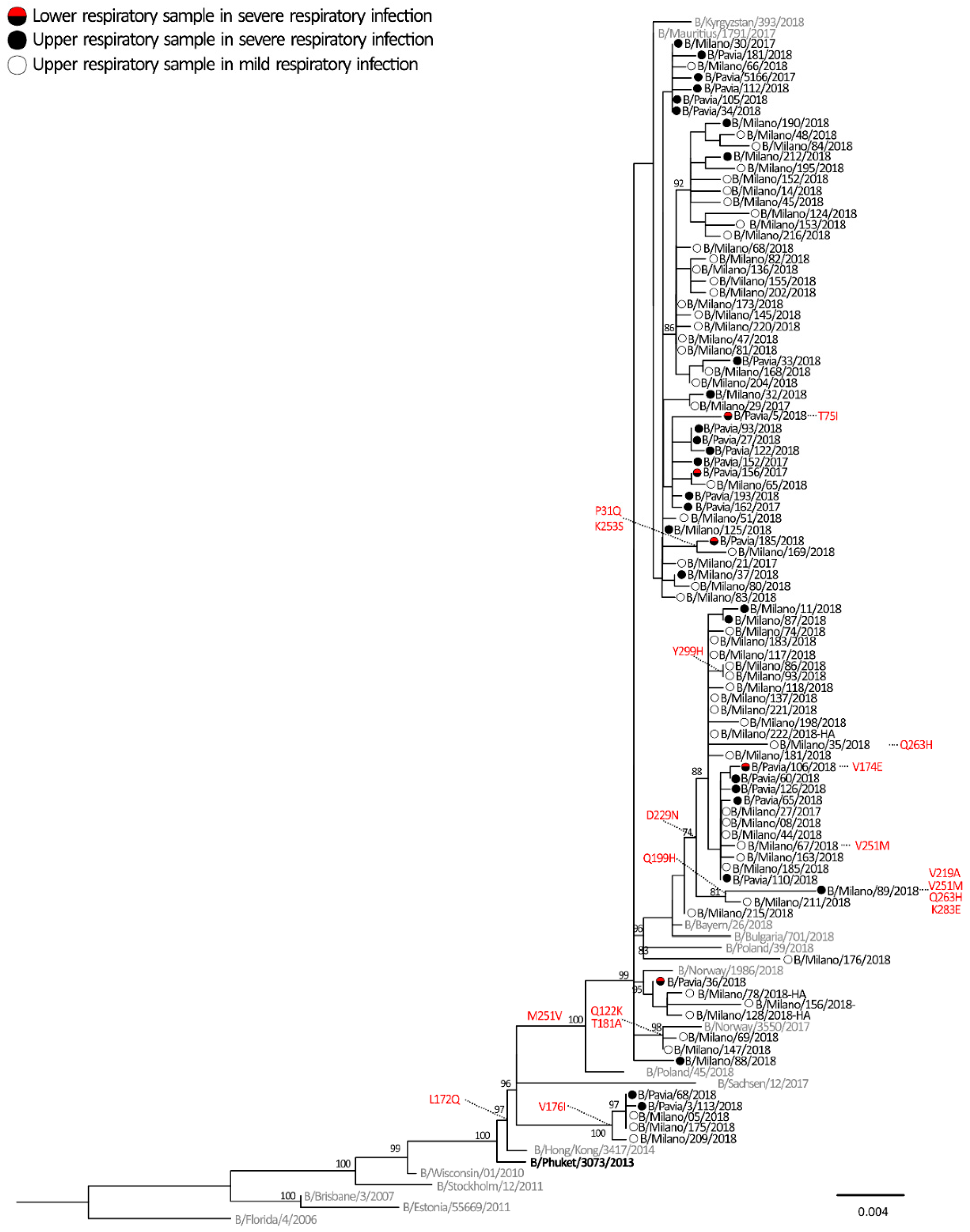
2.3. Phylogenetic Analyses
2.4. Molecular Signatures of Increased Virulence in HA Gene of Influenza virus A and B
2.5. Selective Pressure Analyses
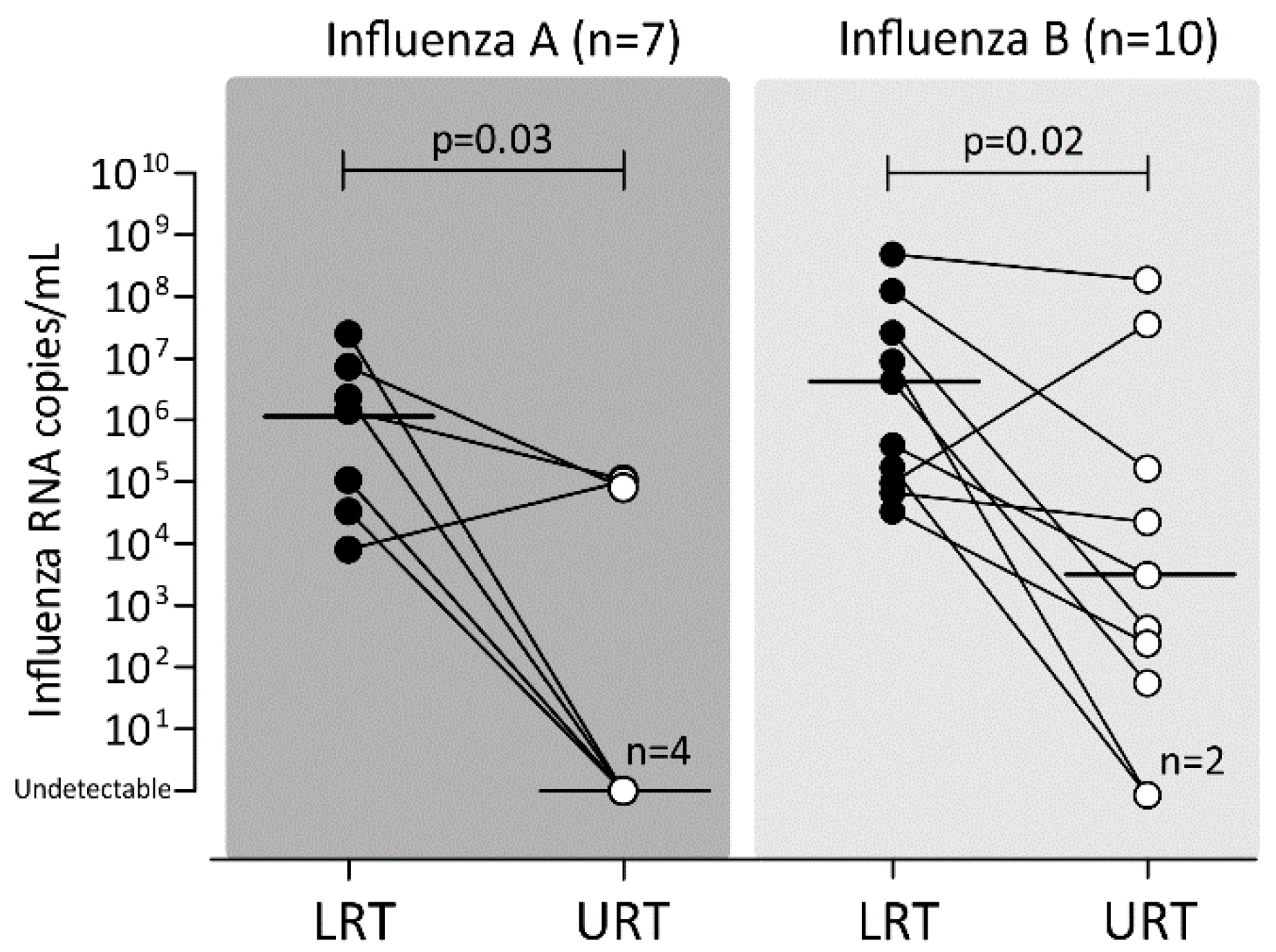
2.6. Viral Load in Paired URT and LRT Samples
3. Discussion
4. Material and Methods
4.1. Study Design
4.2. Ethical Statement
4.3. Detection and Molecular Characterization of Influenza Viruses
4.4. Sequencing of Influenza HA Gene
4.5. Phylogenetic Analyses
4.6. Selective Pressure
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 390, 697–708. [Google Scholar] [CrossRef]
- Word Health Organization. Influenza. Available online: https://www.who.int/influenza/en/ (accessed on 15 March 2019).
- LaRussa, P. Pandemic novel 2009 H1N1 influenza: What have we learned? Semin. Respir. Crit. Care Med. 2011, 32, 393–399. [Google Scholar] [CrossRef]
- European Center for Disease Control (ECDC). Surveillance of severe disease due to influenza in Europe. Available online: http://www.ecdc.europa.eu/en/healthtopics/seasonal_influenza/Documents/1201_ECDC_concept_paper_Surveillance_of_severe_disease_due_to_influenza_in_Europe.pdf (accessed on 25 March 2019).
- European Center for Disease Control (ECDC). Seasonal influenza vaccination in Europe, 2007–2008 to 2014–2015. Available online: https://ecdc.europa.eu/sites/portal/files/documents/influenza-vaccination-2007%E2%80%932008-to-2014%E2%80%932015.pdf (accessed on 25 March 2019).
- Treanor, J. What Happens Next Depends on What Happened First. Clin. Infect. Dis. 2018, 67, 1533–1534. [Google Scholar] [CrossRef]
- Baldanti, F.; Campanini, G.; Piralla, A.; Rovida, F.; Braschi, A.; Mojoli, F.; Iotti, G.; Belliato, M.; Conaldi, PG.; Arcadipane, A.; et al. Severe outcome of influenza A/H1N1/09v infection associated with 222G/N polymorphisms in the haemagglutinin: A multicentre study. Clin. Microbiol. Infect. 2011, 17, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Childs, R.A.; Matrosovich, T.; Wharton, S.; Palma, A.S.; Chai, W.; Daniels, R.; Gregory, V.; Uhlendorff, J.; Kiso, M.; et al. Altered receptor specificity and cell tropism of D222G hemagglutinin mutants isolated from fatal cases of pandemic A(H1N1) 2009 influenza virus. J. Virol. 2010, 84, 12069–12074. [Google Scholar] [CrossRef]
- Lee, H.K.; Tang, J.W.; Loh, T.P.; Oon, L.L.; Koay, E.S. Predicting clinical severity based on substitutions near epitope A of influenza A/H3N2. Infect. Genet. Evol. 2015, 34, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Baranovich, T.; Vongphrachanh, P.; Ketmayoon, P.; Sisouk, T.; Chomlasack, K.; Khanthamaly, V.; Nguyen, H.T.; Mishin, V.P.; Marjuki, H.; Barnes, J.R.; et al. Antiviral Drug-Resistant Influenza B Viruses Carrying H134N Substitution in Neuraminidase, Laos, February 2016. Emerg. Infect. Dis. 2017, 23, 686–690. [Google Scholar] [CrossRef]
- Fineberg, H.V. Pandemic preparedness and response—Lessons from the H1N1 influenza of 2009. N. Engl. J. Med. 2014, 370, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- European Influenza Surveillance Network (EISN). Available online: https://ecdc.europa.eu/en/about-us/partnerships-and-networks/disease-and-laboratory-networks/eisn (accessed on 25 March 2019).
- European Centre for Disease Prevention and Control. Influenza in Europe, summary of the season 2017–2018. Available online: https://ecdc.europa.eu/en/seasonal-influenza/season-2017-18 (accessed on 25 March 2019).
- Epicentro. Influenza stagionale 2017–2018: Il punto della situazione. Available online: http://www.epicentro.iss.it/problemi/influenza/InfluenzaStagionale2017-18.asp (accessed on 25 March 2019).
- World Health Organization (WHO). Recommended composition of influenza virus vaccines for use in the 2017–2018 northern hemisphere influenza season. Wkly. Epidemiol. Record 2017, 92, 117–128. [Google Scholar]
- Francis Crick Institute—Worldwide Influenza Centre. Report prepared for the WHO annual consultation on the composition of influenza vaccine for the Northern Hemisphere 2018–2019. 19th–21st February 2018. Available online: https://www.crick.ac.uk/sites/default/files/2018-07/crick_feb2018_report_for_the_web.pdf (accessed on 25 March 2019).
- Van Kerkhove, M.D.; Vandemaele, K.A.; Shinde, V.; Jaramillo-Gutierrez, G.; Koukounari, A.; Donnelly, C.A.; Carlino, L.O.; Owen, R.; Paterson, B.; Pelletier, L.; et al. Risk factors for severe outcomes following 2009 influenza A (H1N1) infection: A global pooled analysis. PLoS Med. 2011, 8, e1001053. [Google Scholar] [CrossRef] [PubMed]
- Chutinimitkul, S.; Herfst, S.; Steel, J.; Lowen, A.C.; Ye, J.; van Riel, D.; Schrauwen, E.J.; Bestebroer, T.M.; Koel, B.; Burke, D.F.; et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J. Virol. 2010, 84, 11802–11813. [Google Scholar] [CrossRef] [PubMed]
- Kilander, A.; Rykkvin, R.; Dudman, S.G.; Hungnes, O. Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome, Norway 2009–2010. Euro Surveill. 2010, 15, 19498. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, M.; Katano, H.; Nakajima, N.; Tobiume, M.; Ainai, A.; Sekizuka, T.; Hasegawa, H.; Tashiro, M.; Sasaki, Y.; Arakawa, Y.; et al. Characterization of quasispecies of pandemic 2009 influenza A virus (A/H1N1/2009) by de novo sequencing using a next-generation DNA sequencer. PLoS ONE 2010, 5, e10256. [Google Scholar] [CrossRef]
- Wedde, M.; Wählisch, S.; Wolff, T.; Schweiger, B. Predominance of HA-222D/G polymorphism in influenza A(H1N1)pdm09 viruses associated with fatal and severe outcomes recently circulating in Germany. PLoS ONE 2013, 8, e57059. [Google Scholar] [CrossRef]
- Piralla, A.; Pariani, E.; Rovida, F.; Campanini, G.; Muzzi, A.; Emmi, V.; Iotti, G.A.; Pesenti, A.; Conaldi, P.G.; Zanetti, A.; et al. Segregation of virulent influenza A(H1N1) variants in the lower respiratory tract of critically ill patients during the 2010-2011 seasonal epidemic. PLoS ONE 2011, 6, e28332. [Google Scholar] [CrossRef] [PubMed]
- Selleri, M.; Piralla, A.; Rozera, G.; Giombini, E.; Bartolini, B.; Abbate, I.; Campanini, G.; Rovida, F.; Dossena, L.; Capobianchi, M.R.; et al. Detection of haemagglutinin D222 polymorphisms in influenza A(H1N1)pdm09-infected patients by ultra-deep pyrosequencing. Clin. Microbiol. Infect. 2013, 19, 668–673. [Google Scholar] [CrossRef] [Green Version]
- Piralla, A.; Rovida, F.; Girello, A.; Premoli, M.; Mojoli, F.; Belliato, M.; Braschi, A.; Iotti, G.; Pariani, E.; Bubba, L.; et al. Frequency of respiratory virus infections and next-generation analysis of influenza A/H1N1pdm09 dynamics in the lower respiratory tract of patients admitted to the ICU. PLoS ONE 2017, 12, e0178926. [Google Scholar] [CrossRef] [PubMed]
- McCullers, J.A.; Hayden, F.G. Fatal influenza B infections: Time to reexamine influenza research priorities. J. Infect. Dis. 2012, 205, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, E.; Qiu, X.; Wilson, P.C.; Bahl, J.; Krammer, F. The influenza virus hemagglutinin head evolves faster than the stalk domain. Sci. Rep. 2018, 8, 10432. [Google Scholar] [CrossRef]
- Furuse, Y.; Shimabukuro, K.; Odagiri, T.; Sawayama, R.; Okada, T.; Khandaker, I.; Suzuki, A.; Oshitani, H. Comparison of selection pressures on the HA gene of pandemic (2009) and seasonal human and swine influenza A H1 subtype viruses. Virology 2010, 405, 314–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Ayala, E.; Hill, S.; Chung, J.; Miyai, T.; Kagawa, F.T.; Kirsch, C.M.; Jensen, W.A.; Wehner, J.H.; Mohindra, V.; et al. Diagnosis of 2009 Influenza A H1N1: Diagnostic Utility of Blind Endotracheal Aspirate In Intubated Patients With False Negative Realtime Reverse Transcriptase Polymerase Chain Reaction Assays From Nasopharyngeal Samples. Am. J. Respir. Crit. Care Med. 2010, 181, A2623. [Google Scholar]
- Bogoch, I.I.; Andrews, J.R.; Zachary, K.C.; Hohmann, E.L. Diagnosis of influenza from lower respiratory tract sampling after negative upper respiratory tract sampling. Virulence 2013, 4, 82–84. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Vasoo, S.; Stevens, J.; Schreckenberger, P.; Trenholme, G. Pitfalls in diagnosis of pandemic (novel) A/H1N1 2009 influenza. J. Clin. Microbiol. 2010, 48, 1501–1503. [Google Scholar] [CrossRef]
- Yeh, E.; Luo, R.F.; Dyner, L.; Hong, D.K.; Banaei, N.; Baron, E.J.; Pinsky, B.A. Preferential lower respiratory tract infection in swine-origin 2009 A(H1N1) influenza. Clin. Infect. Dis. 2010, 50, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, J.; Qasmieh, S.; Mounts, A.W.; Alexander, B.; Besselaar, T.; Briand, S.; Brown, C.; Clark, S.; Dueger, E.; Gross, D.; et al. Revision of clinical case definitions: Influenza-like illness and severe acute respiratory infection. Bull World Health Organ. 2018, 96, 122–128. [Google Scholar] [CrossRef]
- Piralla, A.; Lunghi, G.; Percivalle, E.; Viganò, C.; Nasta, T.; Pugni, L.; Mosca, F.; Stronati, M.; Torresani, E.; Baldanti, F. FilmArray® respiratory panel performance in respiratory samples from neonatal care units. Diagn. Microbiol. Infect. Dis. 2014, 79, 183–186. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Global Influenza Surveillance Network. Manual for the Laboratory Diagnosis and Virological Surveillance of Influenza. Available online: https://apps.who.int/iris/bitstream/handle/10665/44518/9789241548090_eng.pdf;jsessionid=26D7BFEE30E142D47EA63936B7BDA53A?sequence=1 (accessed on 29 April 2019).
- Piralla, A.; Lunghi, G.; Ruggiero, L.; Girello, A.; Bianchini, S.; Rovida, F.; Caimmi, S.; Marseglia, G.L.; Principi, N.; Baldanti, F.; et al. Molecular epidemiology of influenza B virus among hospitalized pediatric patients in Northern Italy during the 2015–16 season. PLoS ONE 2017, 12, e0185893. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Delport, W.; Poon, A.F.; Frost, S.D.; Kosakovsky Pond, S.L. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Influenza A/H1N1pdm09 Virus Codon | Influenza B Virus Codon | ||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| SLAC | None | 431, 472, 544 | None | 19 sites |
| FEL | 120, 137, 205, 250 | 472, 544 | 122, 181 | None |
| FUBAR | 137, 222 | 39 sites | 229, 251 | 114 sites |
| MEME | 222 | None | 253 | None |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piralla, A.; Pariani, E.; Giardina, F.; Galli, C.; Sapia, D.; Pellegrinelli, L.; Novazzi, F.; Anselmi, G.; Rovida, F.; Mojoli, F.; et al. Molecular Characterization of Influenza Strains in Patients Admitted to Intensive Care Units during the 2017–2018 Season. Int. J. Mol. Sci. 2019, 20, 2664. https://doi.org/10.3390/ijms20112664
Piralla A, Pariani E, Giardina F, Galli C, Sapia D, Pellegrinelli L, Novazzi F, Anselmi G, Rovida F, Mojoli F, et al. Molecular Characterization of Influenza Strains in Patients Admitted to Intensive Care Units during the 2017–2018 Season. International Journal of Molecular Sciences. 2019; 20(11):2664. https://doi.org/10.3390/ijms20112664
Chicago/Turabian StylePiralla, Antonio, Elena Pariani, Federica Giardina, Cristina Galli, Davide Sapia, Laura Pellegrinelli, Federica Novazzi, Giovanni Anselmi, Francesca Rovida, Francesco Mojoli, and et al. 2019. "Molecular Characterization of Influenza Strains in Patients Admitted to Intensive Care Units during the 2017–2018 Season" International Journal of Molecular Sciences 20, no. 11: 2664. https://doi.org/10.3390/ijms20112664
APA StylePiralla, A., Pariani, E., Giardina, F., Galli, C., Sapia, D., Pellegrinelli, L., Novazzi, F., Anselmi, G., Rovida, F., Mojoli, F., Cereda, D., Senatore, S., & Baldanti, F. (2019). Molecular Characterization of Influenza Strains in Patients Admitted to Intensive Care Units during the 2017–2018 Season. International Journal of Molecular Sciences, 20(11), 2664. https://doi.org/10.3390/ijms20112664