mTOR and Aging: An Old Fashioned Dress

Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Protein Translation
3. Mitochondrial and Oxidative Stress
4. Inflammation
5. Stem Cell Pool Turnover
6. Autophagy
7. Cellular Senescence
8. Aging, mTOR, and Cardiovascular Diseases
9. Aging, mTOR, and the Immune System
10. Aging, mTOR, and Cancer
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations:
| 4EBP1 | (Eukaryotic translation initiation factor) 4E binding protein 1 |
| AKT | Proteine kinase B |
| AMBRA1 | Activating molecule in Beclin-1 regulated autophagy |
| AMP | Adenosinmonophosphate |
| AMPK | Adenosinmonophosphate-activated protein kinase |
| ATP | Adenosintriphosphate |
| Bcl2 | B-cell lymphoma 2 |
| BlyS | B lymphocyte Simulator |
| CAT | Catalase |
| CCR7 | Chemokine receptor 7 |
| CDK | Cyclin-dependent kinase |
| c-Myc | myc-oncogene |
| DCs | Dendritic Cells |
| Flt3L | FMS-like tyrosine 3 kinase ligand |
| GM-CSF | Granulocyte-Macrophage colony-stimulating factor |
| GPx | Glutathione peroxidase |
| HIF1α | Hypoxia-inducible factor alpha |
| HO-1 | Heme oxygenase-1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IDO | Indoleamine 2,3-dioxygenase |
| IFN-γ | Interferon-gamma |
| IGF-1 | Insulin growth factor 1 |
| IL1 | Interleukin 1 |
| IL-12 | Interleukin 12 |
| IL6 | Interleukin 6 |
| IL-8 | Interleukin 8 |
| ILT3 | Immunoglobuline-link transcription 3 |
| ILT4 | Immunoglobuline-link transcription 4 |
| iNOS | Inducible nitric oxide synthase |
| LKB1 | Liver kinase B1 |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated proteine kinase |
| MDSCs | Myeloid derived suppressor cells |
| MHC-II | Major histocompatibility complex class II |
| MIP-1α | Macrophage inflammatory protein 1α |
| MMP9 | Matrix-metalloproteinase |
| mTOR | Mammalian Target of Rapamycin |
| MyD88 | Myeloid differentiation primary response 88 |
| NADPH | Nicotinamide dinucleotide phosphate hydrogen |
| NK | Natural Killer |
| Nox | Nitric oxide |
| Nrf-2 | Nuclear factor erythroid-related factor 2 |
| pDC | Plasmacytoid Dendritic Cells |
| PGC-1 α | Peroxisome proliferator-activated receptor-γ coactivators α |
| PGC-1β | Peroxisome proliferator-activated receptor-γ coactivators β |
| PI3-Kinasi | Phospho insitol 3 kinase |
| PPP | Pentose phosphate pathway |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| S6K | S 6 kinase |
| SASP | Senescence-Associated Secretory Phenotype |
| SIRT4 | Sirtuin 4 |
| SOD | Superoxide dismutase |
| SREBP-1 | Sterol regulatory element binding protein 1 |
| Tbet | T cell transcription factor |
| TGF-β | Transforming growth factor β |
| TLR | Toll like receptor |
| TLR2 | Toll like receptor 2 |
| TLR4 | Toll like receptor 4 |
| TNF-α | Tumor necrosis factor-α |
| Treg | Regulatory T cell |
| TSC1/2 | Tuberous sclerosis complex 1/2 |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VEGF | Vascular endothelial growth factor |
| VSMC | Vascular smooth muscle cell |
References
- Bulterijs, S.; Hull, R.S.; Björk, V.C.; Roy, A.G. It is time to classify biological aging as a disease. Front. Genet. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caplan, A.L. Death as an unnatural process. Why is it wrong to seek a cure for aging? EMBO Rep. 2005, 6, S72–S75. [Google Scholar] [CrossRef] [PubMed]
- Zhavoronkov, A.; Bhullar, B. Classifying aging as a disease in the context of ICD-11. Front. Genet. 2015, 6, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stambler, I. Recognizing Degenerative Aging as a Treatable Medical Condition: Methodology and Policy. Aging Dis. 2017, 8, 583–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faragher, R.G. Should we treat aging as a disease? The consequences and dangers of miscategorisation. Front. Genet. 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blagosklonny, M.V. Disease or not, aging is easily treatable. Ageing 2018, 10, 3067–3078. [Google Scholar] [CrossRef]
- Dilman, V.M.; Revskoy, S.Y.; Golubev, A.G. Neuroendocrine-ontogenetic mechanism of aging: Toward an integrated theory of aging. Int. Rev. Neurobiol. 1986, 28, 89–156. [Google Scholar]
- Dilman, V.M. Four models of medicine: Mechanisms of aging and conditions promoting cancer development. Ann. N. Y. Acad. Sci. 1988, 521, 226–227. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Validation of anti-aging drugs by treating age-related diseases. Aging 2009, 1, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Oh, W.J.; Jacinto, E. mTOR complex two signaling and functions. Cell Cycle 2011, 10, 2305–2318. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and-independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Vellai, T.; Takacs-Vellai, K.; Zhang, Y.; Kovacs, A.L.; Orosz, L.; Müller, F. Genetics: Influence of TOR kinase on lifespan in C. elegans. Nature 2003, 426, 620. [Google Scholar] [CrossRef] [PubMed]
- Kaeberlein, M.; Powers, R.W., 3rd; Steffen, K.K.; Westman, E.A.; Hu, D.; Dang, N.; Kerr, E.O.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005, 310, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Anisimov, V.N.; Zabezhinski, M.A.; Popovich, I.G.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Antoch, M.P.; Blagosklonny, M.V. Rapamycin extends maximal lifespan in cancer-prone mice. Am. J. Pathol. 2010, 176, 2092–2097. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Chantranupong, L.; Keys, H.R.; Wang, T.; Gray, N.S.; Sabatini, D.M. A unifying model for mTORC1 mediated regulation of mRNA translation. Nature 2012, 485, 109–113. [Google Scholar] [CrossRef]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014, 334, 474–483. [Google Scholar] [CrossRef]
- Selman, C.; Tullet, J.M.; Wieser, D.D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase one signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef]
- Conn, C.S.; Qian, S.B. mTOR signaling in protein homeostasis: Less is more? Cell Cycle 2011, 10, 1940–1947. [Google Scholar] [CrossRef]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signaling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Conti, V.; Izzo, V.; Corbi, G.; Russomanno, G.; Manzo, V.; De Lise, F.; Di Donato, A.; Filippelli, A. Antioxidant supplementation in the treatment of aging-associated diseases. Front. Pharmacol. 2016, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Yang, J.; Yang, L. Insights for Oxidative Stress and mTOR Signaling in Myocardial Ischemia/Reperfusion Injury under Diabetes. Oxid. Med. Cell Longev. 2017, 2017, 6437467. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Zheng, P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci. Signal. 2009, 2, 98ra75. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Zhang, H.; Puleston, D.J.; Simon, A.K. Autophagy and Immune Senescence. Trends Mol. Med. 2016, 22, 671–686. [Google Scholar] [CrossRef]
- Fenton, R.A.; Dickson, E.W.; Meyer, T.E.; Dobson, J.G., Jr. Aging reduces the cardioprotective effect of ischemic preconditioning in the rat heart. J. Mol. Cell. Cardiol. 2000, 32, 1371–1375. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signalling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Geroconversion: Irreversible step to cellular senescence. Cell Cycle 2014, 13, 3628–3635. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; di Fagagna, F.D. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Cardiovascular regulatory mechanisms in advanced age. Physiol. Rev. 1993, 73, 413–467. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Schulz, R.; Heusch, G. Loss of cardioprotection with ageing. Cardiovasc. Res. 2009, 83, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Strait, J.B.; Lakatta, E.G. Aging-associated cardiovascular changes and their relationship to heart failure. Heart Fail. Clin. 2012, 8, 143–164. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part I: Aging arteries: A “set up” for vascular disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef]
- Lakatta, E.G. Cardiovascular aging research: The next horizons. J. Am. Geriatr. Soc. 1999, 47, 613–625. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar]
- Judge, S.; Leeuwenburgh, C. Cardiac mitochondrial bioenergetics, oxidative stress and aging. Am. J. Physiol. Cell Physiol. 2007, 292, 1983–1992. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Nadtochiy, S.M.; Brookes, P.S.; Nehrke, K. Ischemic preconditioning: The role of mitochondria and aging. Exp. Gerontol. 2012, 47, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ago, T.; Matsushima, S.; Kuroda, J.; Zablocki, D.; Kitazono, T.; Sadoshima, J. The NADPH oxidase Nox4 and aging in the heart. Aging 2010, 2, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Cesselli, D.; Beltrami, A.P.; D’Aurizio, F.; Marcon, P.; Bergamin, N.; Toffoletto, B.; Pandolfi, M.; Puppato, E.; Marino, L.; Signore, S.; et al. Effects of age and heart failure on human cardiac stem cell function. Am. J. Pathol. 2011, 179, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.S.; de Boer, R.A.; Samani, N.J.; van Veldhuisen, D.J.; van der Harst, P. Telomere biology in heart failure. Eur. J. Heart Fail. 2008, 10, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Miyata, S.; Kawase, Y.; Okada, H.; Maruyama, R.; Fujiwara, H. Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy 2006, 2, 212–214. [Google Scholar] [CrossRef]
- Alvers, A.L.; Fishwick, L.K.; Wood, M.S.; Hu, D.; Chung, H.S.; Dunn, W.A., Jr.; Aris, J.P. Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae. Aging Cell 2009, 8, 353–369. [Google Scholar] [CrossRef]
- Hars, E.S.; Qi, H.; Ryazanov, A.G.; Jin, S.; Cai, L.; Hu, C.; Liu, L.F. Autophagy regulates ageing in C. elegans. Autophagy 2007, 3, 93–95. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Taneike, M.; Yamaguchi, O.; Nakai, A.; Hikoso, S.; Takeda, T.; Mizote, I.; Oka, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; et al. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010, 6, 600–606. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Riera, C.E.; Merkwirth, C.; Filho, C.D.D.M.; Dillin, A. Signaling networks determining life span. Annu. Rev. Biochem. 2016, 85, 35–64. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.O.; Lee, Y.K.; Kim, J.M.; Yoon, G. From cell senescence to age-related diseases: Differential mechanisms of action of senescence-associated secretory phenotypes. BMB Rep. 2015, 48, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Coppé, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Monticone, R.E.; Lakatta, E.G. Proinflammation of aging central arteries. Gerontology 2014, 60, 519–529. [Google Scholar] [CrossRef]
- Paneni, F.; Diaz Cañestro, C.; Libby, P.; Lüscher, T.F.; Camici, G.G. The aging cardiovascular system: Understanding it at the cellular and clinical levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef]
- Fourcade, S.; Ferrer, I.; Pujol, A. Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: A paradigm for axonal degeneration. Free Radic. Biol. Med. 2015, 88, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Takayuki, H. Klotho upregulation by rapamycin protects against vascular disease in CKD. Kidney Int. 2015, 88, 660–662. [Google Scholar] [Green Version]
- Vervloet, M.G.; Adema, A.Y.; Larsson, T.E.; Massy, Z.A. The role of klotho on vascular calcification and endothelial function in chronic Kidney disease. Semin. Nephrol. 2014, 34, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Fedarko, N.S. Biology of Aging and Frailty. Clin. Geriatr. Med. 2011, 27, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Hepper, H.J.; Sieber, C.; Walger, P.; Bahrmann, P.; Sigler, K. Infections in the elderly. Crit. Care Clin. 2013, 29, 757–774. [Google Scholar] [CrossRef] [PubMed]
- Larbi, A.; Franceschi, C.; Mazzatti, D.; Solana, R.; Wikby, A.; Pawelec, G. Aging of the immune system as a prognostic factor for human longevity. Physiology 2008, 23, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Licastro, F.; Candore, G.; Lio, D.; Porcellini, E.; Colonna-Romano, G.; Franceschi, C. Innate immunity and inflammation in ageing: A key for understanding age-related diseases. Immun. Ageing 2005, 2, 8. [Google Scholar] [CrossRef]
- Dorshkind, K.; Swain, S. Age-associated declines in immune system development and function: Causes, consequences, and reversal. Curr. Opin. Immunol. 2009, 21, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Effros, R.B. Telomerase induction in T cells: A cure for aging and disease? Exp. Gerontol. 2007, 42, 416–420. [Google Scholar] [CrossRef] [Green Version]
- McKay, D.; Jameson, J. Kidney transplantation and the ageing immune system. Nat. Rev. Nephrol. 2012, 8, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.; Ikematsu, H.; Yamaji, K.; Nishimura, M.; Kashiwagi, S.; Hayashi, J. Age-related accumulation of Ig VH gene somatic mutations in peripheral B cells from aged humans. Clin. Exp. Immunol. 2003, 133, 59–66. [Google Scholar] [CrossRef]
- Mahbub, S.; Brubaker, A.L.; Kovacs, E.J. Aging of the Innate Immune System: An Update. Curr. Immunol. Rev. 2011, 7, 104–115. [Google Scholar] [CrossRef]
- Colonna-Romano, G.; Aquino, A.; Bulati, M.; Di Lorenzo, G.; Listì, F.; Vitello, S. Memory B cell subpopulations in the aged. Rejuvenation Res. 2006, 9, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Scialabba, G.; Candore, G.; Lio, D. B cells in the aged: CD27, CD5, and CD40 expression. Mech. Ageing Dev. 2003, 124, 389–393. [Google Scholar] [CrossRef]
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Vitello, S.; Lio, D.; Candore, G. B cell immunosenescence in the elderly and in centenarians. Rejuvenation Res. 2008, 1, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Appay, V.; Campisi, J.; Frasca, D.; Fülöp, T.; Sauce, D.; Larbi, A.; Weinberger, B.; Cossarizza, A. Aging of the immune system: Focus on inflammation and vaccination. Eur. J. Immunol. 2016, 46, 2286–2301. [Google Scholar] [CrossRef] [PubMed]
- Stout, R.D.; Suttles, J. Immunosenescence and macrophage functional plasticity: Dysregulation of macrophage function by age-associated microenvironmental changes. Immunol. Rev. 2005, 205, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Herrero, C.; Marqués, L.; Lloberas, J.; Celada, A. IFN-γ-dependent transcription of MHC class IIIA is impaired in macrophages from aged mice. J. Clin. Investig. 2001, 107, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.; Nagarkatti, M.; Nagarkatti, P.S.; Subbarao, B.; Udhayakumar, V. Macrophages but not B cells from aged mice are defective in stimulating autoreactive T cells in vitro. Mech. Ageing Dev. 1990, 52, 107–124. [Google Scholar] [CrossRef]
- Zissel, G.; Schlaak, M.; Muller-Quernheim, J. Age-related decrease in accessory cell function of human alveolar macrophages. J. Investig. Med. 1999, 47, 51–56. [Google Scholar]
- Caro-Maldonado, A.; Gerriets, V.A.; Rathmell, J.C. Matched and mismatched metabolic fuels in lymphocyte function. Semin. Immunol. 2012, 24, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–304. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The Transcription Factor Myc Controls Metabolic Reprogramming upon T Lymphocyte Activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, J.D.; Lerner, C.G.; Schwartz, R.H. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J. Immunol. 1999, 162, 2775–2784. [Google Scholar] [PubMed]
- Zheng, Y.; Delgoffe, G.M.; Meyer, C.F.; Chan, W.; Powell, J.D. Anergic T cells are metabolically anergic. J. Immunol. 2009, 183, 6095–6101. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, L.V.; Finlay, D.; Feijoo, C.; Cornish, G.H.; Gray, A.; Ager, A.; Okkenhaug, K.; Hagenbeek, T.J.; Spits, H.; Cantrell, D.A. Phosphatidylinositol-3-OH-kinase and nutrient-sensing mTOR pathways control T lymphocyte trafficking. Nat. Immunol. 2008, 9, 513–521. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Gudapati, P.; Dragovic, S.; Spencer, C.; Joyce, S.; Killeen, N.; Magnuson, M.A.; Boothby, M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 2010, 32, 743–753. [Google Scholar] [CrossRef]
- Wing, K.; Sakaguchi, S. Regulatory T cells exert checks and balances on self-tolerance and autoimmunity. Nat. Immunol. 2010, 11, 7–13. [Google Scholar] [CrossRef]
- Guillonneau, C.; Picarda, E.; Anegon, I. CD8+ regulatory T cells in solid organ transplantation. Curr. Opin. Organ. Transplant. 2010, 15, 751–756. [Google Scholar] [CrossRef]
- Dai, Z.; Zhang, S.; Xie, Q.; Wu, S.; Su, J.; Li, S.; Xu, Y.; Li, X.C. Natural CD8+CD122+ T cells are more potent in suppression of allograft rejection than CD4+CD25+ regulatory T cells. Am. J. Transplant. 2014, 14, 39–48. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.G. Rapamycin selectively expands CD4+ CD25+ FoxP3+ regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Whiteside, T.L.; Knights, A.; Bergmann, C.; Knuth, A.; Zippelius, A. Selective survival of naturally occurring human CD4+ CD25+ Foxp3+ regulatory T cells cultured with rapamycin. J. Immunol. 2007, 178, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Kopf, H.; de la Rosa, G.M.; Howard, O.M.; Chen, X. Rapamycin inhibits differentiation of Th17 cells and promotes generation of FoxP3+ T regulatory cells. Int. Immunopharmacol. 2007, 7, 1819–1824. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.R.; Li, Q.; Odunsi, K.; Shrikant, P.A. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 2010, 32, 67–78. [Google Scholar] [CrossRef]
- Kapsenberg, M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 2003, 3, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Taner, T.; Hackstein, H.; Wang, Z.; Morelli, A.E.; Thomson, A.W. Rapamycin-treated, alloantigen pulsed host dendritic cells induce ag-specific T cell regulation and prolong graft survival. Am. J. Transplant. 2005, 5, 228–236. [Google Scholar] [CrossRef]
- Turnquist, H.R.; Raimondi, G.; Zahorchak, A.F.; Fischer, R.T.; Wang, Z.; Thomson, A.W. Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J. Immunol. 2007, 178, 7018–7031. [Google Scholar] [CrossRef]
- Hackstein, H.; Taner, T.; Zahorchak, A.F.; Morelli, A.E.; Logar, A.J.; Gessner, A.; Thomson, A.W. Rapamycin inhibits IL-4 induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 2003, 101, 4457–4463. [Google Scholar] [CrossRef]
- Waskow, C.; Liu, K.; Darrasse-Jèze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef]
- Woltman, A.M.; de Fijter, J.W.; Kamerling, S.W.; van Der Kooij, S.W.; Paul, L.C.; Daha, M.R.; van Kooten, C. Rapamycin induces apoptosis in monocyte and CD34-derived dendritic cells but not in monocytes and macrophages. Blood 2001, 98, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Sordi, V.; Bianchi, G.; Buracchi, C.; Mercalli, A.; Marchesi, F.; D’Amico, G.; Yang, C.H.; Luini, W.; Vecchi, A.; Mantovani, A.; et al. Differential effects of immunosuppressive drugs on chemokine receptor CCR7 in human monocyte derived dendritic cells: Selective up-regulation by rapamycin. Transplantation 2006, 82, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Pontrelli, P.; Infante, B.; Gigante, M.; Netti, G.S.; Ranieri, E.; Grandaliano, G.; Gesualdo, L. Rapamycin induces ILT3(high)ILT4(high) dendritic cells promoting a new immunoregulatory pathway. Kidney Int. 2014, 85, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloidderived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Lees, J.R.; Azimzadeh, A.M.; Bromberg, J.S. Myeloid derived suppressor cells in transplantation. Curr. Opin. Immunol. 2011, 23, 692–967. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, A.C.; Zea, A.H.; Hernandez, C.; Rodriguez, P.C. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin. Cancer Res. 2007, 13, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin–cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef]
- Mencacci, A.; Montagnoli, C.; Bacci, A.; Cenci, E.; Pitzurra, L.; Spreca, A.; Kopf, M.; Sharpe, A.H.; Romani, L. CD80+ Gr-1+ myeloid cells inhibit development of antifungal Th1 immunity in mice with candidiasis. J. Immunol. 2002, 169, 3180–3190. [Google Scholar] [CrossRef]
- Delano, M.J.; Scumpia, P.O.; Weinstein, J.S.; Coco, D.; Nagaraj, S.; Kelly-Scumpia, K.M.; O’Malley, K.A.; Wynn, J.L.; Antonenko, S.; Al-Quran, S.Z.; et al. MyD88-dependent expansion of an immature GR-1+ CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 2007, 204, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Voisin, M.B.; Buzoni-Gatel, D.; Bout, D.; Velge-Roussel, F. Both expansion of regulatory GR1+ CD11b+ myeloid cells and anergy of T lymphocytes participate in hyporesponsiveness of the lung associated immune system during acute toxoplasmosis. Infect. Immun. 2004, 72, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Bingisser, R.M.; Tilbrook, P.A.; Holt, P.G.; Kees, U.R. Macrophage-derived nitric oxide regulates T-cell activation via reversible disruption of the Jak3/ STAT5 signaling pathway. J. Immunol. 1998, 160, 5729–5734. [Google Scholar] [PubMed]
- Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar]
- De Wilde, V.; Van Rompaey, N.; Hill, M.; Lebrun, J.F.; Lemaître, P.; Lhommé, F.; Kubjak, C.; Vokaer, B.; Oldenhove, G.; Charbonnier, L.M.; et al. Endotoxin-induced myeloid-derived suppressor cells inhibit alloimmune responses via heme oxygenase-1. Am. J. Transplant. 2009, 9, 2034–2047. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef]
- Baban, B.; Chandler, P.R.; Johnson, B.A., 3rd; Huang, L.; Li, M.; Sharpe, M.L.; Francisco, L.M.; Sharpe, A.H.; Blazar, B.R.; Munn, D.H.; et al. Physiologic control of IDO competence in splenic dendritic cells. J. Immunol. 2011, 187, 2329–2335. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. The role of myeloid-derived suppressor cells (MDSC) in the inflammaging process. Ageing Res. Rev. 2018, 48, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhao, Y.; Wang, H.; Li, Y.; Shao, L.; Wang, R.; Lu, J.; Yang, Z.; Wang, J.; Zhao, Y. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci. Rep. 2016, 6, 20250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannick, J.B.; Morris, M.; Hockey, H.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melief, C.J. Cancer immunotherapy by dendritic cells. Immunity 2008, 29, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gabrilovich, D.I. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008, 68, 2561–2563. [Google Scholar] [CrossRef]
- Myers, C.E.; Mirza, N.N.; Lustgarten, J. Immunity, cancer and aging: Lessons from mouse models. Aging Dis. 2011, 2, 512–523. [Google Scholar]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef]
- Pawelec, G.; Lustgarten, J.; Ruby, C.; Gravekamp, C. Impact of aging on cancer immunity and immunotherapy. Cancer Immunol. Immunother. 2009, 58, 1907–1908. [Google Scholar] [CrossRef]
- Deng, Y.; Chan, S.S.; Chang, S. Telomere dysfunction and tumour suppression: The senescence connection. Nat. Rev. Cancer 2008, 8, 450–458. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Von Zglinicki, T.; Saretzki, G.; Ladhoff, J.; d’Adda di Fagagna, F.; Jackson, S.P. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 2005, 126, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Burnet, M. Cancer—A biological approach. I. the processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Provinciali, M.; Barucca, A.; Cardelli, M.; Marchegiani, F.; Pierpaoli, E. Inflammation, aging, and cancer vaccines. Biogerontology 2010, 11, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27, S43–S51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yecies, J.L.; Manning, B.D. Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res. 2011, 71, 2815–2820. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Bi, Y.; Shen, B.; Yang, H.; Zhang, Y.; Wang, X.; Liu, H.; Lu, Y.; Liao, J.; Chen, X.; et al. SIRT1 Limits the Function and Fate of Myeloid-Derived Suppressor Cells in Tumors by Orchestrating HIF-1-α Dependent Glycolysis. Cancer Res. 2014, 74, 727–737. [Google Scholar] [CrossRef]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. MTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. The mTOR complex 2 is required for the development of prostate cancer induced by P-ten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef]
- Qi, W.X.; Huang, Y.J.; Yao, Y.; Shen, Z.; Min, D.L. Incidence and Risk of Treatment-Related Mortality with mTOR Inhibitors Everolimus and Temsirolimus in Cancer Patients: A Meta-Analysis. PLoS ONE 2013, 8, e65166. [Google Scholar] [CrossRef]
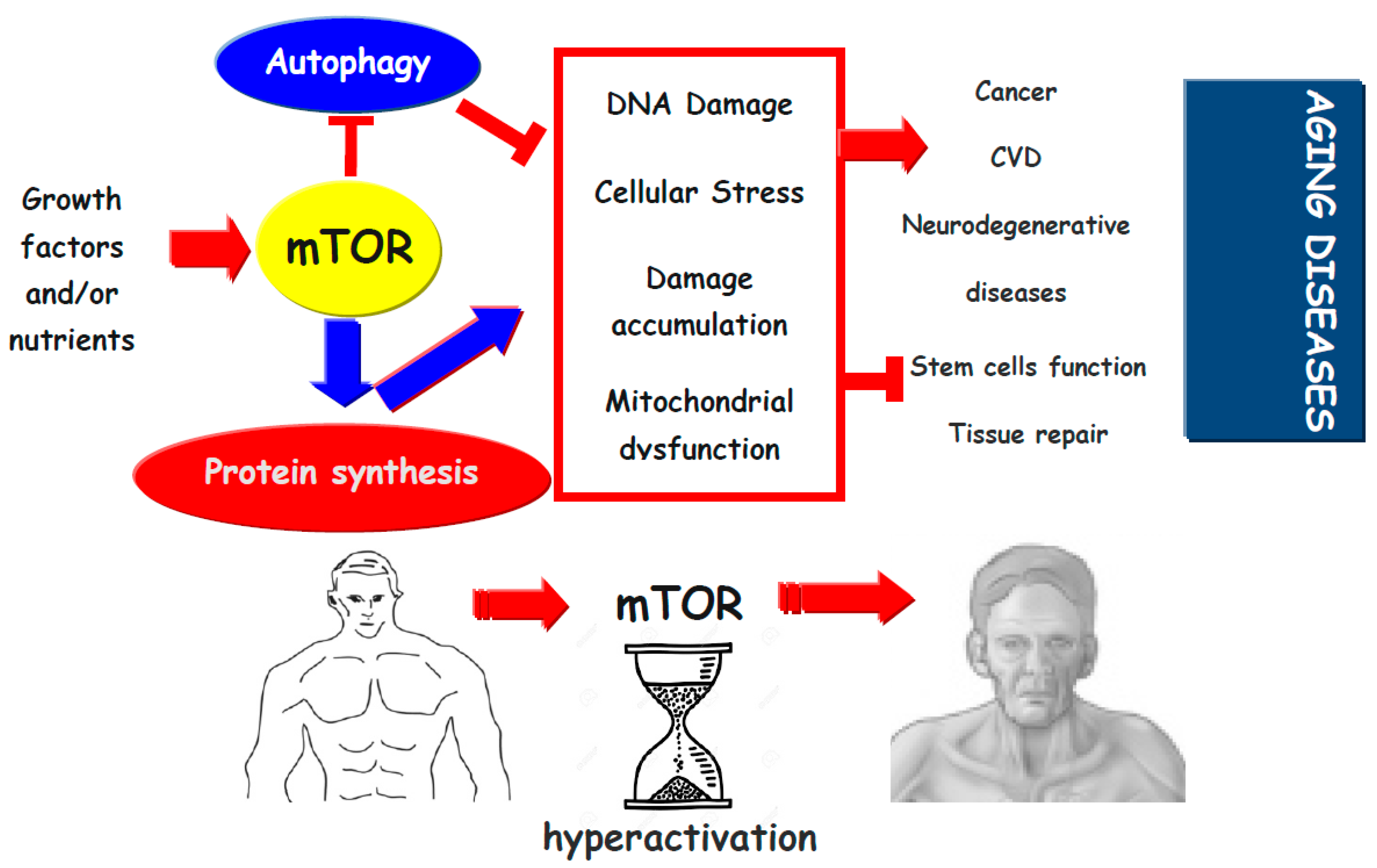
 (blue and red) indicates activation,
(blue and red) indicates activation,  indicates inhibition.
(blue and red) indicates activation, indicates inhibition.
indicates inhibition.
(blue and red) indicates activation, indicates inhibition.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stallone, G.; Infante, B.; Prisciandaro, C.; Grandaliano, G. mTOR and Aging: An Old Fashioned Dress. Int. J. Mol. Sci. 2019, 20, 2774. https://doi.org/10.3390/ijms20112774
Stallone G, Infante B, Prisciandaro C, Grandaliano G. mTOR and Aging: An Old Fashioned Dress. International Journal of Molecular Sciences. 2019; 20(11):2774. https://doi.org/10.3390/ijms20112774
Chicago/Turabian StyleStallone, Giovanni, Barbara Infante, Concetta Prisciandaro, and Giuseppe Grandaliano. 2019. "mTOR and Aging: An Old Fashioned Dress" International Journal of Molecular Sciences 20, no. 11: 2774. https://doi.org/10.3390/ijms20112774
APA StyleStallone, G., Infante, B., Prisciandaro, C., & Grandaliano, G. (2019). mTOR and Aging: An Old Fashioned Dress. International Journal of Molecular Sciences, 20(11), 2774. https://doi.org/10.3390/ijms20112774