The Evolving Role of CD8+CD28? Immunosenescent T Cells in Cancer Immunology

Abstract
:1. Introduction
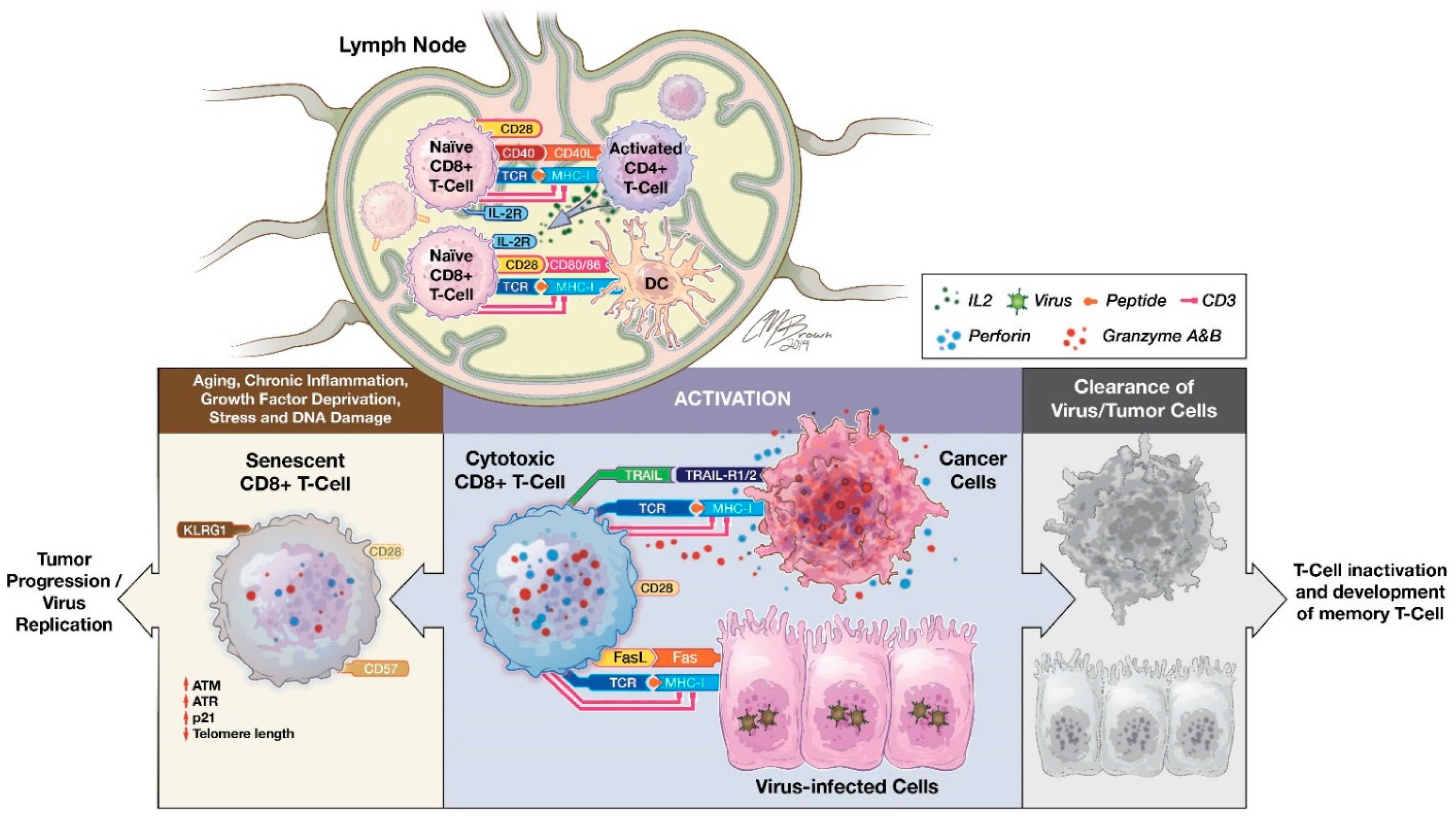
2. Role of CD8+ T Cells in Cancer Immunology
3. CD28 Costimulatory Receptor
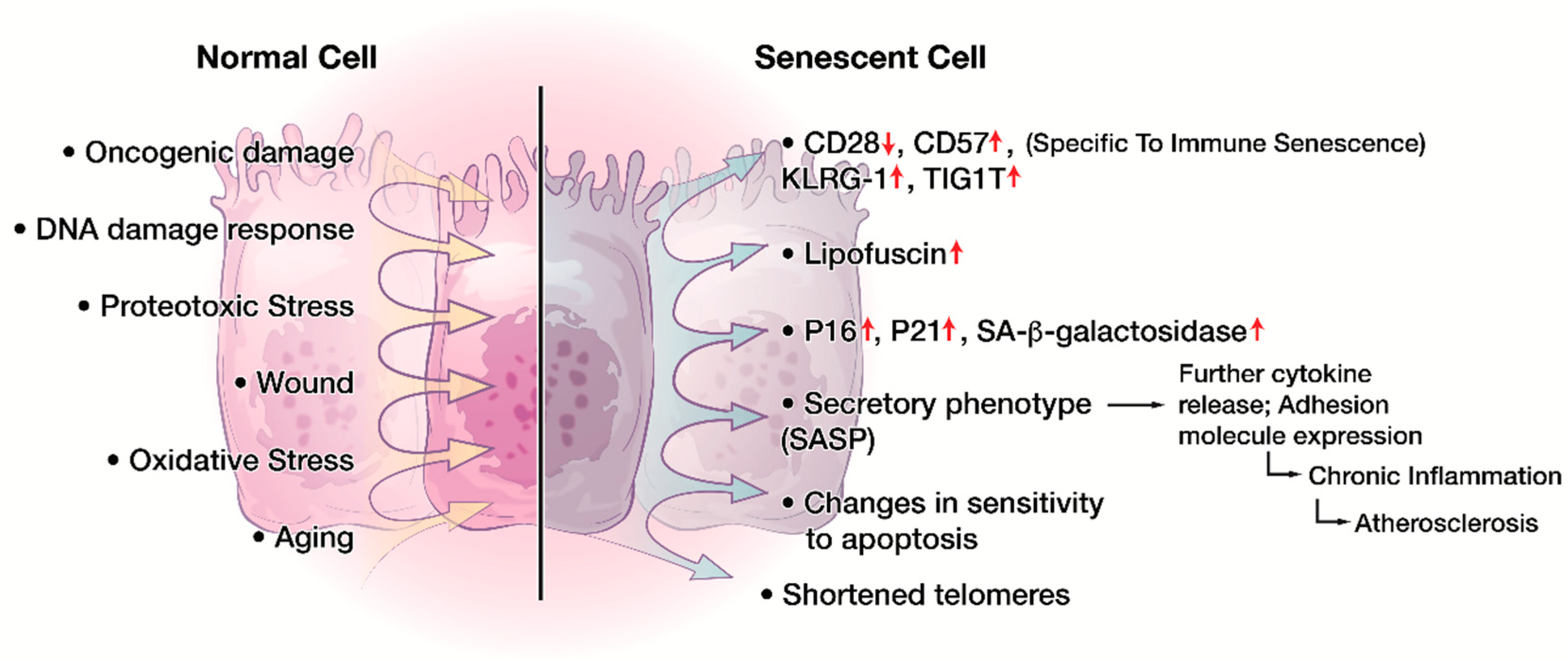
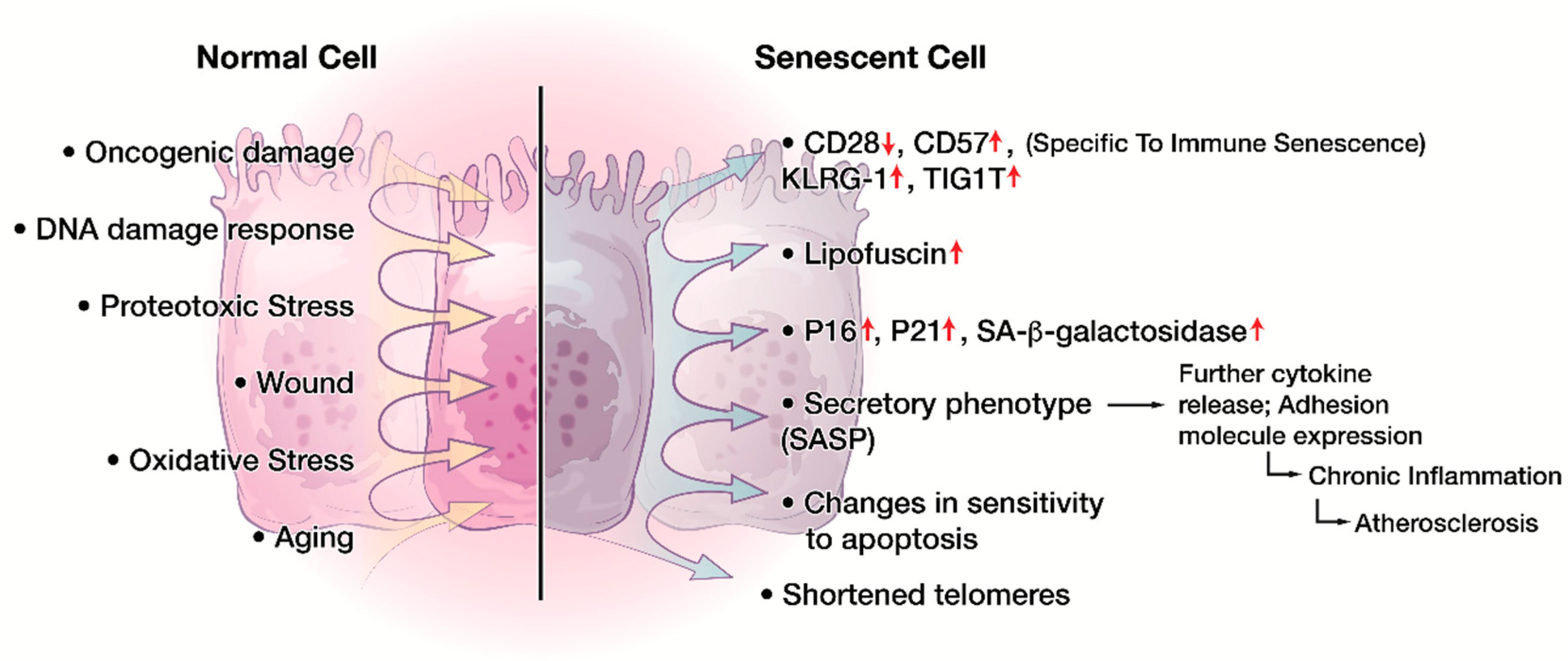
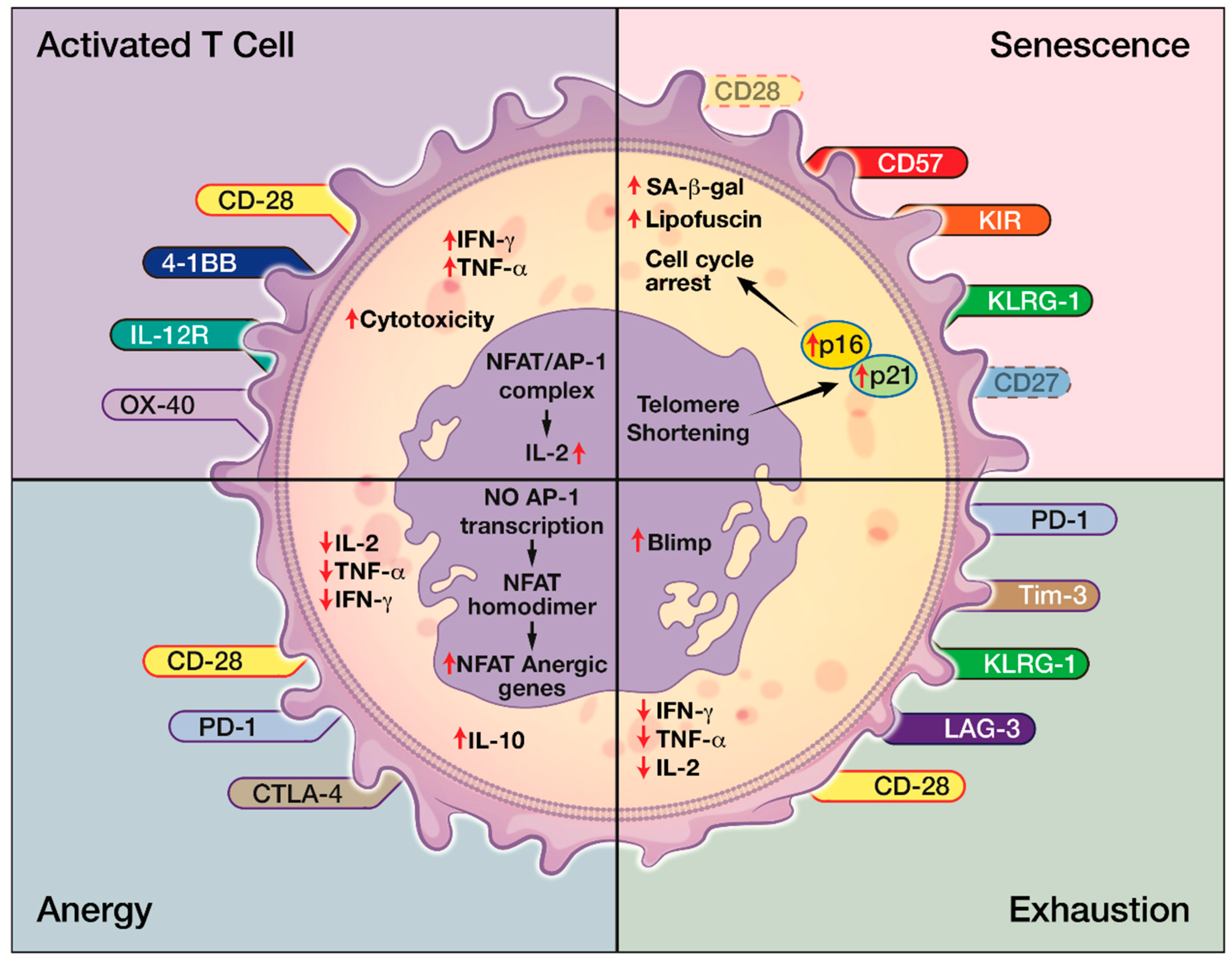
4. Cellular Senescence in the Immune System
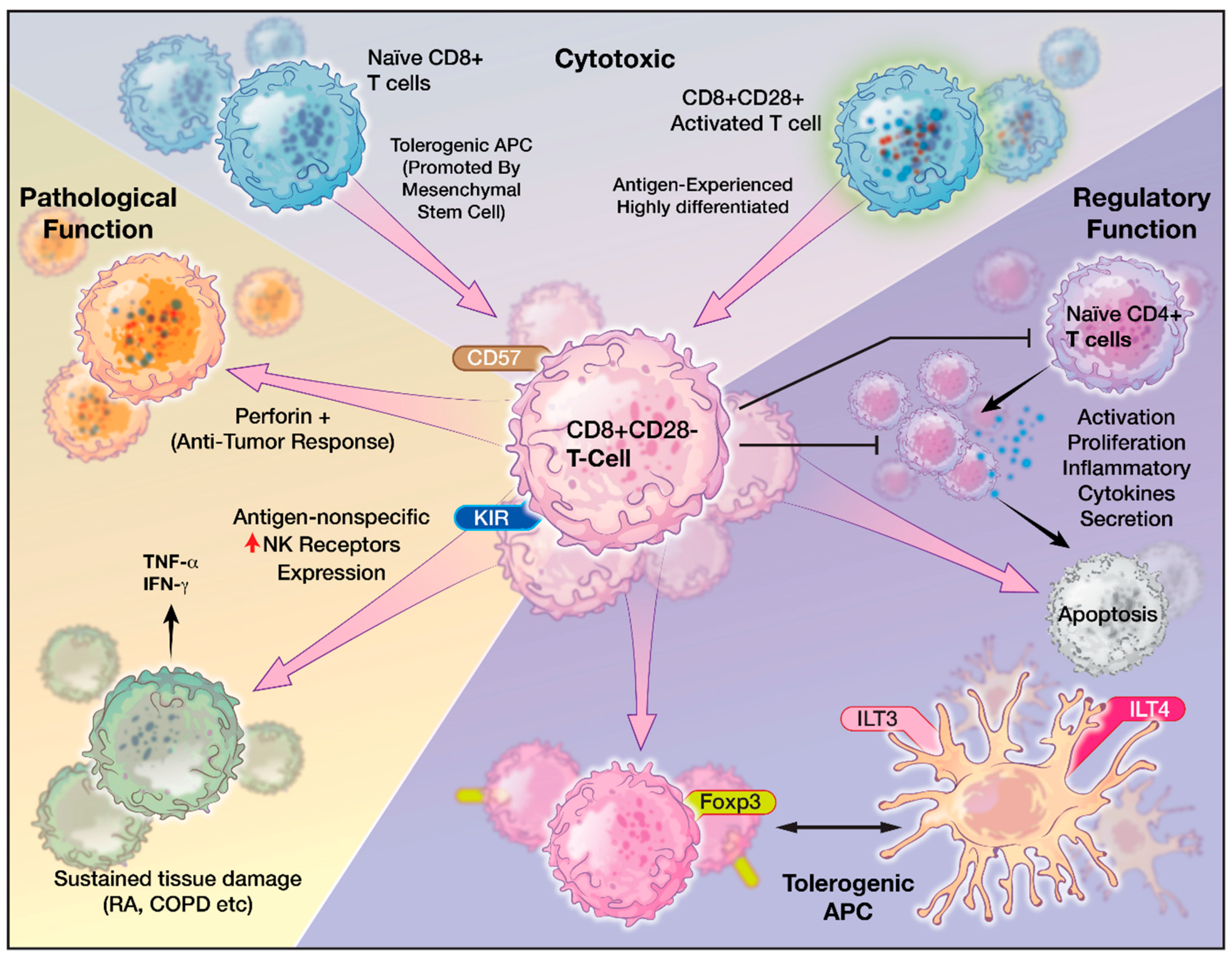
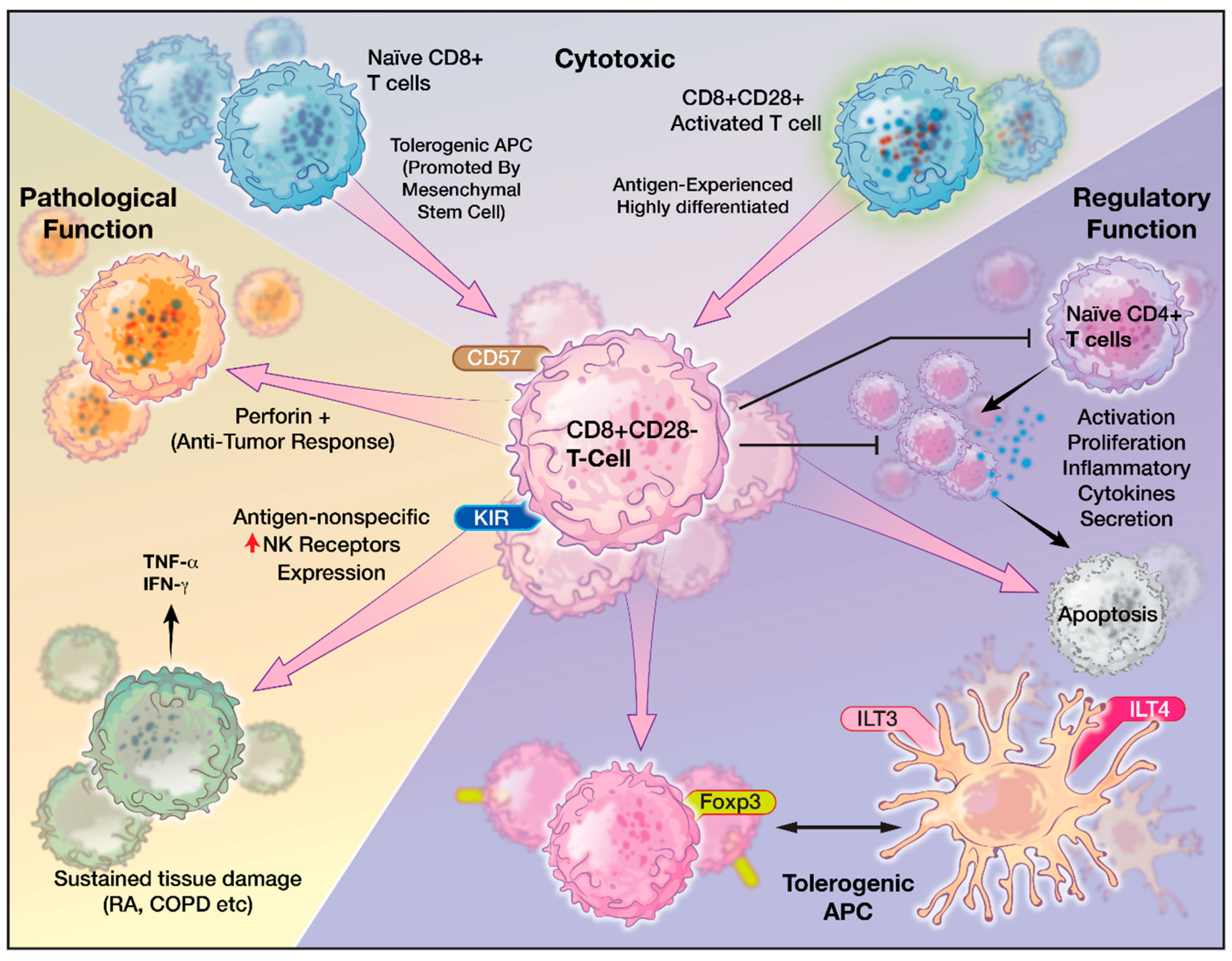
5. Role of CD8+CD28− T Cells
6. Characteristics of CD8+CD28− Senescent T cells
7. CD8+CD28− T cells in Pathologic Conditions
8. CD8+CD28− T cells and Cancer
9. CD8+CD28− T cells and Glioblastoma
10. Implications of CD8+CD28− T Cells for the Future of Immunotherapy
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cells |
| CAR | Chimeric antigen receptor |
| CMV | Cytomegalovirus |
| CTL | Cytotoxic T-lymphocytes |
| DC | Dendritic cells |
| GBM | Glioblastoma |
| NK | Natural killer |
| SA-β-Gal | Senescence-associated-β-galactosidase |
| TCR | T cell receptor |
| Th | T helper cells |
| TME | Tumor microenvironment |
| Tregs | T- regulatory cells |
References
- Palucka, A.K.; Coussens, L.M. The basis of oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Lee, H.H.; Lee, H.J.; Choi, W.S.; Lee, J.; Kim, H.S. Natural killer cells as a promising therapeutic target for cancer immunotherapy. Arch. Pharm. Res. 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.M.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic immunity is required for effective cancer immunotherapy. Cell 2017, 168, 487–502.e15. [Google Scholar] [CrossRef] [PubMed]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kuhnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? Bmc Med. 2016, 14, 73. [Google Scholar] [CrossRef]
- Moreira, A.; Gross, S.; Kirchberger, M.C.; Erdmann, M.; Schuler, G.; Heinzerling, L. Senescence markers: Predictive for response to checkpoint inhibitors. Int. J. Cancer 2019, 144, 1147–1150. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase i cohorts of checkmate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; de Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 randomized phase 3 study evaluating the efficacy and safety of nivolumab vs bevacizumab in patients with recurrent glioblastoma: Checkmate 143. Neuro Oncol. 2017, 19, iii21. [Google Scholar] [CrossRef]
- Powles, T.; Duran, I.; van der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castellano, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (imvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-l1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Crespo, J.; Sun, H.; Welling, T.H.; Tian, Z.; Zou, W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T cell dysfunction in cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef]
- Aguilera, M.O.; Delgui, L.R.; Romano, P.S.; Colombo, M.I. Chronic infections: A possible scenario for autophagy and senescence cross-talk. Cells 2018, 7, 162. [Google Scholar] [CrossRef]
- Aberg, J.A. Aging, inflammation, and hiv infection. Top. Antivir. Med. 2012, 20, 101–105. [Google Scholar]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef]
- Warren, J.A.; Clutton, G.; Goonetilleke, N. Harnessing CD8(+) T cells under hiv antiretroviral therapy. Front. Immunol. 2019, 10, 291. [Google Scholar] [CrossRef]
- Schwartz, R.H. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Q.; Xiang, A.P. Cd8+CD28- T cells: Not only age-related cells but a subset of regulatory T cells. Cell. Mol. Immunol. 2018, 15, 734–736. [Google Scholar] [CrossRef]
- Filaci, G.; Fenoglio, D.; Fravega, M.; Ansaldo, G.; Borgonovo, G.; Traverso, P.; Villaggio, B.; Ferrera, A.; Kunkl, A.; Rizzi, M.; et al. CD8+ CD28- T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J. Immunol. 2007, 179, 4323–4334. [Google Scholar] [CrossRef]
- Borthwick, N.J.; Lowdell, M.; Salmon, M.; Akbar, A.N. Loss of CD28 expression on CD8(+) T cells is induced by IL-2 receptor gamma chain signalling cytokines and type i ifn, and increases susceptibility to activation-induced apoptosis. Int. Immunol. 2000, 12, 1005–1013. [Google Scholar] [CrossRef]
- Vallejo, A.N. CD28 extinction in human T cells: Altered functions and the program of T-cell senescence. Immunol. Rev. 2005, 205, 158–169. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Babala, N.; Melief, C.J.M.; Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8(+) cytotoxic t lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Singer, A.; Adoro, S.; Park, J.H. Lineage fate and intense debate: Myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat. Rev. Immunol. 2008, 8, 788–801. [Google Scholar] [CrossRef]
- Germain, R.N. T-cell development and the CD4-CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Arosa, F.A.; Esgalhado, A.J.; Padrao, C.A.; Cardoso, E.M. Divide, conquer, and sense: Cd8(+)CD28(-) T cells in perspective. Front. Immunol. 2016, 7, 665. [Google Scholar] [CrossRef]
- Fearon, D.T.; Carr, J.M.; Telaranta, A.; Carrasco, M.J.; Thaventhiran, J.E. The rationale for the il-2-independent generation of the self-renewing central memory CD8+ T cells. Immunol. Rev. 2006, 211, 104–118. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef]
- Reading, J.L.; Galvez-Cancino, F.; Swanton, C.; Lladser, A.; Peggs, K.S.; Quezada, S.A. The function and dysfunction of memory CD8(+) T cells in tumor immunity. Immunol. Rev. 2018, 283, 194–212. [Google Scholar] [CrossRef]
- Ara, A.; Ahmed, K.A.; Xiang, J. Multiple effects of CD40-CD40l axis in immunity against infection and cancer. Immuno Targets Ther. 2018, 7, 55–61. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Darcy, P.K. Gene-engineered T cells for cancer therapy. Nat. Rev. Cancer 2013, 13, 525–541. [Google Scholar] [CrossRef]
- Gottschalk, C.; Mettke, E.; Kurts, C. The role of invariant natural killer T cells in dendritic cell licensing, cross-priming, and memory CD8(+) T cell generation. Front. Immunol. 2015, 6, 379. [Google Scholar] [CrossRef]
- Wu, R.C.; Hwu, P.; Radvanyi, L.G. New insights on the role of CD8(+)CD57(+) T-cells in cancer. Oncoimmunology 2012, 1, 954–956. [Google Scholar] [CrossRef]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.; Ellis, I.O.; Green, A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef]
- Yang, Z.Q.; Yang, Z.Y.; Zhang, L.D.; Ping, B.; Wang, S.G.; Ma, K.S.; Li, X.W.; Dong, J.H. Increased liver-infiltrating CD8+FOXP3+ regulatory T cells are associated with tumor stage in hepatocellular carcinoma patients. Hum. Immunol. 2010, 71, 1180–1186. [Google Scholar] [CrossRef]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef]
- Ling, A.; Edin, S.; Wikberg, M.L.; Oberg, A.; Palmqvist, R. The intratumoural subsite and relation of CD8(+) and FOXP3(+) T lymphocytes in colorectal cancer provide important prognostic clues. Br. J. Cancer 2014, 110, 2551–2559. [Google Scholar] [CrossRef]
- Liu, L.; Zhao, G.; Wu, W.; Rong, Y.; Jin, D.; Wang, D.; Lou, W.; Qin, X. Low intratumoral regulatory T cells and high peritumoral CD8(+) T cells relate to long-term survival in patients with pancreatic ductal adenocarcinoma after pancreatectomy. Cancer Immunol. Immunother. 2016, 65, 73–82. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef]
- Huang, Y.; Ma, C.; Zhang, Q.; Ye, J.; Wang, F.; Zhang, Y.; Hunborg, P.; Varvares, M.A.; Hoft, D.F.; Hsueh, E.C.; et al. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget 2015, 6, 17462–17478. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Lin, J.; Qiao, G.; Xu, Y.; Zou, H. Differential regulation and function of tumor-infiltrating T cells in different stages of breast cancer patients. Tumour Biol. 2015, 36, 7907–7913. [Google Scholar] [CrossRef]
- Watanabe, Y.; Katou, F.; Ohtani, H.; Nakayama, T.; Yoshie, O.; Hashimoto, K. Tumor-infiltrating lymphocytes, particularly the balance between CD8(+) T cells and CCR4(+) regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2010, 109, 744–752. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 costimulation: From mechanism to therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef]
- Zumerle, S.; Molon, B.; Viola, A. Membrane rafts in T cell activation: A spotlight on CD28 costimulation. Front. Immunol. 2017, 8, 1467. [Google Scholar] [CrossRef]
- Porciello, N.; Grazioli, P.; Campese, A.F.; Kunkl, M.; Caristi, S.; Mastrogiovanni, M.; Muscolini, M.; Spadaro, F.; Favre, C.; Nunès, J.A.; et al. A non-conserved amino acid variant regulates differential signalling between human and mouse CD28. Nat. Commun. 2018, 9, 1080. [Google Scholar] [CrossRef] [Green Version]
- Bour-Jordan, H.; Esensten, J.H.; Martinez-Llordella, M.; Penaranda, C.; Stumpf, M.; Bluestone, J.A. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol. Rev. 2011, 241, 180–205. [Google Scholar] [CrossRef]
- Zhang, Q.; Vignali, D.A. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Zeng, Y.; Thistlethwaite, J.R.; Montag, A.; Brady, W.; Gibson, M.G.; Linsley, P.S.; Bluestone, J.A. Long-term survival of xenogeneic pancreatic islet grafts induced by CTLA4IG. Science 1992, 257, 789–792. [Google Scholar] [CrossRef]
- Blair, H.A.; Deeks, E.D. Abatacept: A review in rheumatoid arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef]
- Vincenti, F.; Rostaing, L.; Grinyo, J.; Rice, K.; Steinberg, S.; Gaite, L.; Moal, M.C.; Mondragon-Ramirez, G.A.; Kothari, J.; Polinsky, M.S.; et al. Belatacept and long-term outcomes in kidney transplantation. N. Engl. J. Med. 2016, 374, 333–343. [Google Scholar] [CrossRef]
- Ford, M.L. T cell cosignaling molecules in transplantation. Immunity 2016, 44, 1020–1033. [Google Scholar] [CrossRef]
- Attanasio, J.; Wherry, E.J. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity 2016, 44, 1052–1068. [Google Scholar] [CrossRef]
- Callahan, M.K.; Postow, M.A.; Wolchok, J.D. Targeting T cell co-receptors for cancer therapy. Immunity 2016, 44, 1069–1078. [Google Scholar] [CrossRef]
- Suntharalingam, G.; Perry, M.R.; Ward, S.; Brett, S.J.; Castello-Cortes, A.; Brunner, M.D.; Panoskaltsis, N. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006, 355, 1018–1028. [Google Scholar] [CrossRef]
- Tyrsin, D.; Chuvpilo, S.; Matskevich, A.; Nemenov, D.; Romer, P.S.; Tabares, P.; Hunig, T. From TGN1412 to TAB08: The return of CD28 superagonist therapy to clinical development for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 45–48. [Google Scholar]
- Hunig, T. The storm has cleared: Lessons from the CD28 superagonist TGN1412 trial. Nat. Rev. Immunol. 2012, 12, 317–318. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Aptamers as targeted therapeutics: Current potential and challenges. Nat. Rev. Drug Discov. 2017, 16, 440. [Google Scholar] [CrossRef]
- Lozano, T.; Soldevilla, M.M.; Casares, N.; Villanueva, H.; Bendandi, M.; Lasarte, J.J.; Pastor, F. Targeting inhibition of FOXP3 by a CD28 2’-fluro oligonucleotide aptamer conjugated to p60-peptide enhances active cancer immunotherapy. Biomaterials 2016, 91, 73–80. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Villanueva, H.; Casares, N.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; Lopez-Diaz de Cerio, A.; Pastor, F. MRP1-CD28 bi-specific oligonucleotide aptamers: Target costimulation to drug-resistant melanoma cancer stem cells. Oncotarget 2016, 7, 23182–23196. [Google Scholar] [CrossRef]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, checkmate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase I trial protocol. BMJ Open 2016, 6, e013904. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Wakefield, A.; Ghazi, A.; Ashoori, A.; Diouf, O.; Gerken, C.; Landi, D.; et al. Autologous HER2 cmv bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a phase I trial. J. Immunother. Cancer 2015, 3, O11. [Google Scholar] [CrossRef]
- Cabo, M.; Offringa, R.; Zitvogel, L.; Kroemer, G.; Muntasell, A.; Galluzzi, L. Trial watch: Immunostimulatory monoclonal antibodies for oncological indications. Oncoimmunology 2017, 6, e1371896. [Google Scholar] [CrossRef]
- Emerson, D.A.; Redmond, W.L. Overcoming tumor-induced immune suppression: From relieving inhibition to providing costimulation with T cell agonists. BioDrugs 2018, 32, 221–231. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and icos. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef]
- Pratico, E.D.; Sullenger, B.A.; Nair, S.K. Identification and characterization of an agonistic aptamer against the T cell costimulatory receptor, OX40. Nucleic Acid Ther. 2013, 23, 35–43. [Google Scholar] [CrossRef]
- Weigelin, B.; Bolanos, E.; Teijeira, A.; Martinez-Forero, I.; Labiano, S.; Azpilikueta, A.; Morales-Kastresana, A.; Quetglas, J.I.; Wagena, E.; Sanchez-Paulete, A.R.; et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc. Natl. Acad. Sci. USA 2015, 112, 7551–7556. [Google Scholar] [CrossRef] [Green Version]
- Coe, D.; Begom, S.; Addey, C.; White, M.; Dyson, J.; Chai, J.G. Depletion of regulatory T cells by anti-gitr mab as a novel mechanism for cancer immunotherapy. Cancer Immunol. Immunother. 2010, 59, 1367–1377. [Google Scholar] [CrossRef]
- Schaer, D.A.; Budhu, S.; Liu, C.; Bryson, C.; Malandro, N.; Cohen, A.; Zhong, H.; Yang, X.; Houghton, A.N.; Merghoub, T.; et al. Gitr pathway activation abrogates tumor immune suppression through loss of regulatory T cell lineage stability. Cancer Immunol. Res. 2013, 1, 320–331. [Google Scholar] [CrossRef]
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 2014, 12, 36. [Google Scholar] [CrossRef]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the icos pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Long, E.O. Cellular senescence induced by CD158D reprograms natural killer cells to promote vascular remodeling. Proc. Natl. Acad. Sci. USA 2012, 109, 20596–20601. [Google Scholar] [CrossRef]
- Howcroft, T.K.; Campisi, J.; Louis, G.B.; Smith, M.T.; Wise, B.; Wyss-Coray, T.; Augustine, A.D.; McElhaney, J.E.; Kohanski, R.; Sierra, F. The role of inflammation in age-related disease. Aging 2013, 5, 84–93. [Google Scholar] [CrossRef] [Green Version]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and senotherapeutics: A new field in cancer therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Petrakis, T.G.; Komseli, E.S.; Papaioannou, M.; Vougas, K.; Polyzos, A.; Myrianthopoulos, V.; Mikros, E.; Trougakos, I.P.; Thanos, D.; Branzei, D.; et al. Exploring and exploiting the systemic effects of deregulated replication licensing. Semin. Cancer Biol. 2016, 37–38, 3–15. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Alcorta, D.A.; Xiong, Y.; Phelps, D.; Hannon, G.; Beach, D.; Barrett, J.C. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4A) in replicative senescence of normal human fibroblasts. Proc. Natl. Acad. Sci. USA 1996, 93, 13742–13747. [Google Scholar] [CrossRef]
- Jurk, D.; Wang, C.; Miwa, S.; Maddick, M.; Korolchuk, V.; Tsolou, A.; Gonos, E.S.; Thrasivoulou, C.; Saffrey, M.J.; Cameron, K.; et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 2012, 11, 996–1004. [Google Scholar] [CrossRef]
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495. [Google Scholar] [CrossRef] [Green Version]
- Aird, K.M.; Zhang, R. Detection of senescence-associated heterochromatin foci (SAHF). Methods Mol. Biol. 2013, 965, 185–196. [Google Scholar]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Munoz-Espin, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Immunosenescence: The potential role of myeloid-derived suppressor cells (MDSC) in age-related immune deficiency. Cell. Mol. Life Sci. 2019, 76, 1901–1918. [Google Scholar] [CrossRef]
- Akbar, A.N.; Fletcher, J.M. Memory T cell homeostasis and senescence during aging. Curr. Opin. Immunol. 2005, 17, 480–485. [Google Scholar] [CrossRef]
- Swain, S.; Clise-Dwyer, K.; Haynes, L. Homeostasis and the age-associated defect of CD4 T cells. Semin. Immunol. 2005, 17, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Singhal, S.K.; Roder, J.C.; Duwe, A.K. Suppressor cells in immunosenescence. Fed. Proc. 1978, 37, 1245–1252. [Google Scholar]
- Strioga, M.; Pasukoniene, V.; Characiejus, D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology 2011, 134, 17–32. [Google Scholar] [CrossRef]
- Vallejo, A.N.; Weyand, C.M.; Goronzy, J.J. T-cell senescence: A culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol. Med. 2004, 10, 119–124. [Google Scholar] [CrossRef]
- Eck, S.C.; Chang, D.; Wells, A.D.; Turka, L.A. Differential down-regulation of CD28 by B7-1 and B7-2 engagement. Transplantation 1997, 64, 1497–1499. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4(+) T cells in CD8(+) T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]
- Weng, N.P.; Akbar, A.N.; Goronzy, J. CD28(-) T cells: Their role in the age-associated decline of immune function. Trends Immunol. 2009, 30, 306–312. [Google Scholar] [CrossRef]
- Qin, L.; Jing, X.; Qiu, Z.; Cao, W.; Jiao, Y.; Routy, J.P.; Li, T. Aging of immune system: Immune signature from peripheral blood lymphocyte subsets in 1068 healthy adults. Aging 2016, 8, 848–859. [Google Scholar] [CrossRef]
- Valenzuela, H.F.; Effros, R.B. Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin. Immunol. 2002, 105, 117–125. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef]
- Paillard, F.; Sterkers, G.; Vaquero, C. Transcriptional and post-transcriptional regulation of tcr, CD4 and CD8 gene expression during activation of normal human T lymphocytes. EMBO J. 1990, 9, 1867–1872. [Google Scholar] [CrossRef]
- Mou, D.; Espinosa, J.; Lo, D.J.; Kirk, A.D. CD28 negative T cells: Is their loss our gain? Am. J. Transpl. 2014, 14, 2460–2466. [Google Scholar] [CrossRef]
- Tarazona, R.; DelaRosa, O.; Alonso, C.; Ostos, B.; Espejo, J.; Pena, J.; Solana, R. Increased expression of NK cell markers on T lymphocytes in aging and chronic activation of the immune system reflects the accumulation of effector/senescent T cells. Mech. Ageing Dev. 2000, 121, 77–88. [Google Scholar] [CrossRef]
- Seyda, M.; Elkhal, A.; Quante, M.; Falk, C.S.; Tullius, S.G. T cells going innate. Trends Immunol. 2016, 37, 546–556. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of nk cells and T cells by NKG2D, a receptor for stress-inducible mica. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Verneris, M.R.; Karimi, M.; Baker, J.; Jayaswal, A.; Negrin, R.S. Role of NKG2D signaling in the cytotoxicity of activated and expanded CD8+ T cells. Blood 2004, 103, 3065–3072. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, K.; Perez, C.; Rojas, L.B.P.; Burke, B.; Guevara-Patino, J.A. Functions of NKG2D in CD8(+) T cells: An opportunity for immunotherapy. Cell. Mol. Immunol. 2018, 15, 470–479. [Google Scholar] [CrossRef]
- Yi, H.S.; Kim, S.Y.; Kim, J.T.; Lee, Y.S.; Moon, J.S.; Kim, M.; Kang, Y.E.; Joung, K.H.; Lee, J.H.; Kim, H.J.; et al. T-cell senescence contributes to abnormal glucose homeostasis in humans and mice. Cell Death Dis. 2019, 10, 249. [Google Scholar] [CrossRef]
- Onyema, O.O.; Decoster, L.; Njemini, R.; Forti, L.N.; Bautmans, I.; De Waele, M.; Mets, T. Shifts in subsets of CD8+ T-cells as evidence of immunosenescence in patients with cancers affecting the lungs: An observational case-control study. BMC Cancer 2015, 15, 1016. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Karandikar, N.J.; Betts, M.R.; Ambrozak, D.R.; Hill, B.J.; Crotty, L.E.; Casazza, J.P.; Kuruppu, J.; Migueles, S.A.; Connors, M.; et al. Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101, 2711–2720. [Google Scholar] [CrossRef]
- Gunturi, A.; Berg, R.E.; Forman, J. Preferential survival of CD8 T and NK cells expressing high levels of CD94. J. Immunol. 2003, 170, 1737–1745. [Google Scholar] [CrossRef]
- Appay, V.; Nixon, D.F.; Donahoe, S.M.; Gillespie, G.M.; Dong, T.; King, A.; Ogg, G.S.; Spiegel, H.M.; Conlon, C.; Spina, C.A.; et al. Hiv-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000, 192, 63–75. [Google Scholar] [CrossRef]
- Kiniry, B.E.; Hunt, P.W.; Hecht, F.M.; Somsouk, M.; Deeks, S.G.; Shacklett, B.L. Differential expression of CD8(+) T cell cytotoxic effector molecules in blood and gastrointestinal mucosa in HIV-1 infection. J. Immunol. 2018, 200, 1876–1888. [Google Scholar] [CrossRef]
- Reuter, M.A.; Del Rio Estrada, P.M.; Buggert, M.; Petrovas, C.; Ferrando-Martinez, S.; Nguyen, S.; Sada Japp, A.; Ablanedo-Terrazas, Y.; Rivero-Arrieta, A.; Kuri-Cervantes, L.; et al. HIV-specific CD8(+) T cells exhibit reduced and differentially regulated cytolytic activity in lymphoid tissue. Cell Rep. 2017, 21, 3458–3470. [Google Scholar] [CrossRef]
- Hodge, G.; Hodge, S. Steroid resistant CD8(+)CD28(null) nkt-like pro-inflammatory cytotoxic cells in chronic obstructive pulmonary disease. Front. Immunol. 2016, 7, 617. [Google Scholar] [CrossRef]
- Cortesini, R.; LeMaoult, J.; Ciubotariu, R.; Cortesini, N.S. Cd8+CD28- t suppressor cells and the induction of antigen-specific, antigen-presenting cell-mediated suppression of th reactivity. Immunol. Rev. 2001, 182, 201–206. [Google Scholar] [CrossRef]
- Plaumann, J.; Engelhardt, M.; Awwad, M.H.S.; Echchannaoui, H.; Amman, E.; Raab, M.S.; Hillengass, J.; Halama, N.; Neuber, B.; Muller-Tidow, C.; et al. IL-10 inducible CD8(+) regulatory T-cells are enriched in patients with multiple myeloma and impact the generation of antigen-specific T-cells. Cancer Immunol. Immunother. 2018, 67, 1695–1707. [Google Scholar] [CrossRef]
- Geng, L.; Liu, J.; Huang, J.; Lin, B.; Yu, S.; Shen, T.; Wang, Z.; Yang, Z.; Zhou, L.; Zheng, S. A high frequency of CD8(+)CD28(-) t-suppressor cells contributes to maintaining stable graft function and reducing immunosuppressant dosage after liver transplantation. Int. J. Med. Sci. 2018, 15, 892–899. [Google Scholar] [CrossRef]
- Vieyra-Lobato, M.R.; Vela-Ojeda, J.; Montiel-Cervantes, L.; Lopez-Santiago, R.; Moreno-Lafont, M.C. Description of CD8(+) regulatory T lymphocytes and their specific intervention in graft-versus-host and infectious diseases, autoimmunity, and cancer. J. Immunol. Res 2018, 2018, 3758713. [Google Scholar] [CrossRef]
- Manavalan, J.S.; Rossi, P.C.; Vlad, G.; Piazza, F.; Yarilina, A.; Cortesini, R.; Mancini, D.; Suciu-Foca, N. High expression of ILT3 and ILT4 is a general feature of tolerogenic dendritic cells. Transpl. Immunol. 2003, 11, 245–258. [Google Scholar] [CrossRef]
- Ye, J.; Huang, X.; Hsueh, E.C.; Zhang, Q.; Ma, C.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Human regulatory T cells induce T-lymphocyte senescence. Blood 2012, 120, 2021–2031. [Google Scholar] [CrossRef]
- Sun, Z.; Zhong, W.; Lu, X.; Shi, B.; Zhu, Y.; Chen, L.; Zhang, G.; Zhang, X. Association of graves’ disease and prevalence of circulating IFN-γ-producing CD28(-) T cells. J. Clin. Immunol. 2008, 28, 464–472. [Google Scholar] [CrossRef]
- Maly, K.; Schirmer, M. The story of CD4+ CD28- T cells revisited: Solved or still ongoing? J. Immunol. Res. 2015, 2015, 348746. [Google Scholar]
- Tulunay, A.; Yavuz, S.; Direskeneli, H.; Eksioglu-Demiralp, E. CD8+CD28-, suppressive T cells in systemic lupus erythematosus. Lupus 2008, 17, 630–637. [Google Scholar] [CrossRef]
- Najafian, N.; Chitnis, T.; Salama, A.D.; Zhu, B.; Benou, C.; Yuan, X.; Clarkson, M.R.; Sayegh, M.H.; Khoury, S.J. Regulatory functions of CD8+CD28- T cells in an autoimmune disease model. J. Clin. Investig. 2003, 112, 1037–1048. [Google Scholar] [CrossRef]
- Manavalan, J.S.; Kim-Schulze, S.; Scotto, L.; Naiyer, A.J.; Vlad, G.; Colombo, P.C.; Marboe, C.; Mancini, D.; Cortesini, R.; Suciu-Foca, N. Alloantigen specific CD8+CD28- FOXP3+ T suppressor cells induce ILT3+ ILT4+ tolerogenic endothelial cells, inhibiting alloreactivity. Int. Immunol. 2004, 16, 1055–1068. [Google Scholar] [CrossRef]
- Chang, C.C.; Ciubotariu, R.; Manavalan, J.S.; Yuan, J.; Colovai, A.I.; Piazza, F.; Lederman, S.; Colonna, M.; Cortesini, R.; Dalla-Favera, R.; et al. Tolerization of dendritic cells by T(s) cells: The crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 2002, 3, 237–243. [Google Scholar] [CrossRef]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain tumor microenvironment and host state: Implications for immunotherapy. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef]
- Wargo, J.A.; Reddy, S.M.; Reuben, A.; Sharma, P. Monitoring immune responses in the tumor microenvironment. Curr. Opin. Immunol. 2016, 41, 23–31. [Google Scholar] [CrossRef] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef]
- Mirzaei, R.; Sarkar, S.; Yong, V.W. T cell exhaustion in glioblastoma: Intricacies of immune checkpoints. Trends Immunol. 2017, 38, 104–115. [Google Scholar] [CrossRef]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015, 17 (Suppl. 7), vii9–vii14. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef]
- Cheng, W.; Fu, D.; Xu, F.; Zhang, Z. Unwrapping the genomic characteristics of urothelial bladder cancer and successes with immune checkpoint blockade therapy. Oncogenesis 2018, 7, 2. [Google Scholar] [CrossRef]
- Lauko, A.; Thapa, B.; Venur, V.A.; Ahluwalia, M.S. Management of brain metastases in the new era of checkpoint inhibition. Curr. Neurol. Neurosci. Rep. 2018, 18, 70. [Google Scholar] [CrossRef]
- Johnson, D.B.; Sullivan, R.J.; Menzies, A.M. Immune checkpoint inhibitors in challenging populations. Cancer 2017, 123, 1904–1911. [Google Scholar] [CrossRef] [Green Version]
- Meloni, F.; Morosini, M.; Solari, N.; Passadore, I.; Nascimbene, C.; Novo, M.; Ferrari, M.; Cosentino, M.; Marino, F.; Pozzi, E.; et al. Foxp3 expressing CD4+ CD25+ and CD8+CD28- T regulatory cells in the peripheral blood of patients with lung cancer and pleural mesothelioma. Hum. Immunol. 2006, 67, 1–12. [Google Scholar] [CrossRef]
- Chen, C.; Chen, D.; Zhang, Y.; Chen, Z.; Zhu, W.; Zhang, B.; Wang, Z.; Le, H. Changes of CD4+CD25+FOXP3+ and CD8+CD28- regulatory T cells in non-small cell lung cancer patients undergoing surgery. Int. Immunopharmacol. 2014, 18, 255–261. [Google Scholar] [CrossRef]
- Casado, J.G.; Soto, R.; DelaRosa, O.; Peralbo, E.; del Carmen Munoz-Villanueva, M.; Rioja, L.; Pena, J.; Solana, R.; Tarazona, R. CD8 T cells expressing nk associated receptors are increased in melanoma patients and display an effector phenotype. Cancer Immunol. Immunother. 2005, 54, 1162–1171. [Google Scholar] [CrossRef]
- Liu, X.; Mo, W.; Ye, J.; Li, L.; Zhang, Y.; Hsueh, E.C.; Hoft, D.F.; Peng, G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat. Commun. 2018, 9, 249. [Google Scholar] [CrossRef]
- Ye, J.; Ma, C.; Hsueh, E.C.; Eickhoff, C.S.; Zhang, Y.; Varvares, M.A.; Hoft, D.F.; Peng, G. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J. Immunol. 2013, 190, 2403–2414. [Google Scholar] [CrossRef]
- Ye, J.; Peng, G. Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology 2015, 4, e994398. [Google Scholar] [CrossRef] [Green Version]
- Lanna, A.; Henson, S.M.; Escors, D.; Akbar, A.N. The kinase p38 activated by the metabolic regulator AMPK and scaffold TAB1 drives the senescence of human T cells. Nat. Immunol. 2014, 15, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Ma, C.; Hsueh, E.C.; Dou, J.; Mo, W.; Liu, S.; Han, B.; Huang, Y.; Zhang, Y.; Varvares, M.A.; et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol. Med. 2014, 6, 1294–1311. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Sayour, E.J.; McLendon, P.; McLendon, R.; De Leon, G.; Reynolds, R.; Kresak, J.; Sampson, J.H.; Mitchell, D.A. Increased proportion of FoxP3+ regulatory T cells in tumor infiltrating lymphocytes is associated with tumor recurrence and reduced survival in patients with glioblastoma. Cancer Immunol. Immunother. 2015, 64, 419–427. [Google Scholar] [CrossRef]
- Fornara, O.; Odeberg, J.; Wolmer Solberg, N.; Tammik, C.; Skarman, P.; Peredo, I.; Stragliotto, G.; Rahbar, A.; Soderberg-Naucler, C. Poor survival in glioblastoma patients is associated with early signs of immunosenescence in the CD4 T-cell compartment after surgery. Oncoimmunology 2015, 4, e1036211. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.F. CD8+ regulatory T cells, their suppressive mechanisms, and regulation in cancer. Hum. Immunol. 2008, 69, 811–814. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Vlad, G.; Cortesini, R.; Suciu-Foca, N. CD8+ T suppressor cells and the ILT3 master switch. Hum. Immunol. 2008, 69, 681–686. [Google Scholar] [CrossRef]
- Ladomersky, E.; Scholtens, D.M.; Kocherginsky, M.; Hibler, E.A.; Bartom, E.T.; Otto-Meyer, S.; Zhai, L.; Lauing, K.L.; Choi, J.; Sosman, J.A.; et al. The coincidence between increasing age, immunosuppression, and the incidence of patients with glioblastoma. Front. Pharmacol. 2019, 10, 200. [Google Scholar] [CrossRef]
- Lamas, A.; Lopez, E.; Carrio, R.; Lopez, D.M. Adipocyte and leptin accumulation in tumor-induced thymic involution. Int. J. Mol. Med. 2016, 37, 133–138. [Google Scholar] [CrossRef]
- Kasakovski, D.; Xu, L.; Li, Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018, 11, 91. [Google Scholar] [CrossRef]
- Short, S.; Fielder, E.; Miwa, S.; von Zglinicki, T. Senolytics and senostatics as adjuvant tumour therapy. EBioMedicine 2019, 41, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The clinical potential of senolytic drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Rueff, J.; Medinger, M.; Heim, D.; Passweg, J.; Stern, M. Lymphocyte subset recovery and outcome after autologous hematopoietic stem cell transplantation for plasma cell myeloma. Biol. Blood Marrow Transpl. 2014, 20, 896–899. [Google Scholar] [CrossRef]
- Farge, D.; Arruda, L.C.; Brigant, F.; Clave, E.; Douay, C.; Marjanovic, Z.; Deligny, C.; Maki, G.; Gluckman, E.; Toubert, A.; et al. Long-term immune reconstitution and T cell repertoire analysis after autologous hematopoietic stem cell transplantation in systemic sclerosis patients. J. Hematol. Oncol. 2017, 10, 21. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates abeta-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Allsopp, R. Telomere length and ipsc re-programming: Survival of the longest. Cell Res. 2012, 22, 614–615. [Google Scholar] [CrossRef]
- Parish, S.T.; Wu, J.E.; Effros, R.B. Sustained CD28 expression delays multiple features of replicative senescence in human CD8 T lymphocytes. J. Clin. Immunol. 2010, 30, 798–805. [Google Scholar] [CrossRef]
- Le Page, A.; Fortin, C.; Garneau, H.; Allard, N.; Tsvetkova, K.; Tan, C.T.; Larbi, A.; Dupuis, G.; Fulop, T. Downregulation of inhibitory SRC homology 2 domain-containing phosphatase-1 (SHP-1) leads to recovery of T cell responses in elderly. Cell Commun. Signal. 2014, 12, 2. [Google Scholar] [CrossRef]
- Karagiannis, P.; Iriguchi, S.; Kaneko, S. Reprogramming away from the exhausted T cell state. Semin. Immunol. 2016, 28, 35–44. [Google Scholar] [CrossRef]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Kaneko, S. In vitro generation of antigen-specific T cells from induced pluripotent stem cells of antigen-specific T cell origin. Methods Mol. Biol. 2016, 1393, 67–73. [Google Scholar]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In vivo fate and activity of second- versus third-generation CD19-specific CAR-T cells in B cell non-hodgkin’s lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Petersen, C.T.; Krenciute, G. Next generation CAR T cells for the immunotherapy of high-grade glioma. Front. Oncol. 2019, 9, 69. [Google Scholar] [CrossRef]
- Sahin, A.; Sanchez, C.; Bullain, S.; Waterman, P.; Weissleder, R.; Carter, B.S. Development of third generation anti-egfrviii chimeric T cells and egfrviii-expressing artificial antigen presenting cells for adoptive cell therapy for glioma. PLoS ONE 2018, 13, e0199414. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Al-Chami, E.; Tormo, A.; Pasquin, S.; Kanjarawi, R.; Ziouani, S.; Rafei, M. Interleukin-21 administration to aged mice rejuvenates their peripheral T-cell pool by triggering de novo thymopoiesis. Aging Cell 2016, 15, 349–360. [Google Scholar] [CrossRef]
- Fan, Y.; Tajima, A.; Goh, S.K.; Geng, X.; Gualtierotti, G.; Grupillo, M.; Coppola, A.; Bertera, S.; Rudert, W.A.; Banerjee, I.; et al. Bioengineering thymus organoids to restore thymic function and induce donor-specific immune tolerance to allografts. Mol. Ther. 2015, 23, 1262–1277. [Google Scholar] [CrossRef]
- Tajima, A.; Pradhan, I.; Trucco, M.; Fan, Y. Restoration of thymus function with bioengineered thymus organoids. Curr. Stem Cell Rep. 2016, 2, 128–139. [Google Scholar] [CrossRef]
- Akbar, A.N.; Henson, S.M. Are senescence and exhaustion intertwined or unrelated processes that compromise immunity? Nat. Rev. Immunol. 2011, 11, 289–295. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Malignancy | Phase | N | Trial Name | Clinical Trial Identifier | Therapeutics | References |
|---|---|---|---|---|---|---|
| Relapsed or Refractory Acute Lymphoblastic Leukemia | 1 | 5 | Chimeric Antigen Receptor (CAR)-Modified T Cell Therapy in Treating Patients with Acute Lymphoblastic Leukemia | NCT02186860 | Third Gen CAR-T cells containing CD28+CD137 | [71] |
| Glioblastoma | 1 | 17 | CMV-Specific Cytotoxic T Lymphocytes Expressing CAR Targeting HER2 in Patients with GBM (HERT-GBM) | NCT01109095 | Second Gen CMV-selected CAR-T cells against HER2 containing CD28.zeta signaling domain | [72] |
| Rheumatoid Arthritis | 1/2 | 18 | Safety, Tolerability, Pharmacodynamics and Efficacy Study of TAB08 in Patients with Rheumatoid Arthritis | NCT01990157 | TAB08 | |
| Solid Neoplasms | 1 | 38 | Dose Escalation Study of TAB08 in Patients with Advanced Solid Neoplasms (TAB08) | NCT03006029 | TAB08 | [73] |
| Systemic Lupus Erythematosus | 2 | 730 | Safety and Efficacy Study of a Biologic to Treat Systemic Lupus Erythematosus | NCT02265744 | Lulizumab pegol (monoclonal antibody against CD28) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huff, W.X.; Kwon, J.H.; Henriquez, M.; Fetcko, K.; Dey, M. The Evolving Role of CD8+CD28? Immunosenescent T Cells in Cancer Immunology. Int. J. Mol. Sci. 2019, 20, 2810. https://doi.org/10.3390/ijms20112810
Huff WX, Kwon JH, Henriquez M, Fetcko K, Dey M. The Evolving Role of CD8+CD28? Immunosenescent T Cells in Cancer Immunology. International Journal of Molecular Sciences. 2019; 20(11):2810. https://doi.org/10.3390/ijms20112810
Chicago/Turabian StyleHuff, Wei X., Jae Hyun Kwon, Mario Henriquez, Kaleigh Fetcko, and Mahua Dey. 2019. "The Evolving Role of CD8+CD28? Immunosenescent T Cells in Cancer Immunology" International Journal of Molecular Sciences 20, no. 11: 2810. https://doi.org/10.3390/ijms20112810
APA StyleHuff, W. X., Kwon, J. H., Henriquez, M., Fetcko, K., & Dey, M. (2019). The Evolving Role of CD8+CD28? Immunosenescent T Cells in Cancer Immunology. International Journal of Molecular Sciences, 20(11), 2810. https://doi.org/10.3390/ijms20112810