Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges

 ,
,  , ,
, ,
Abstract
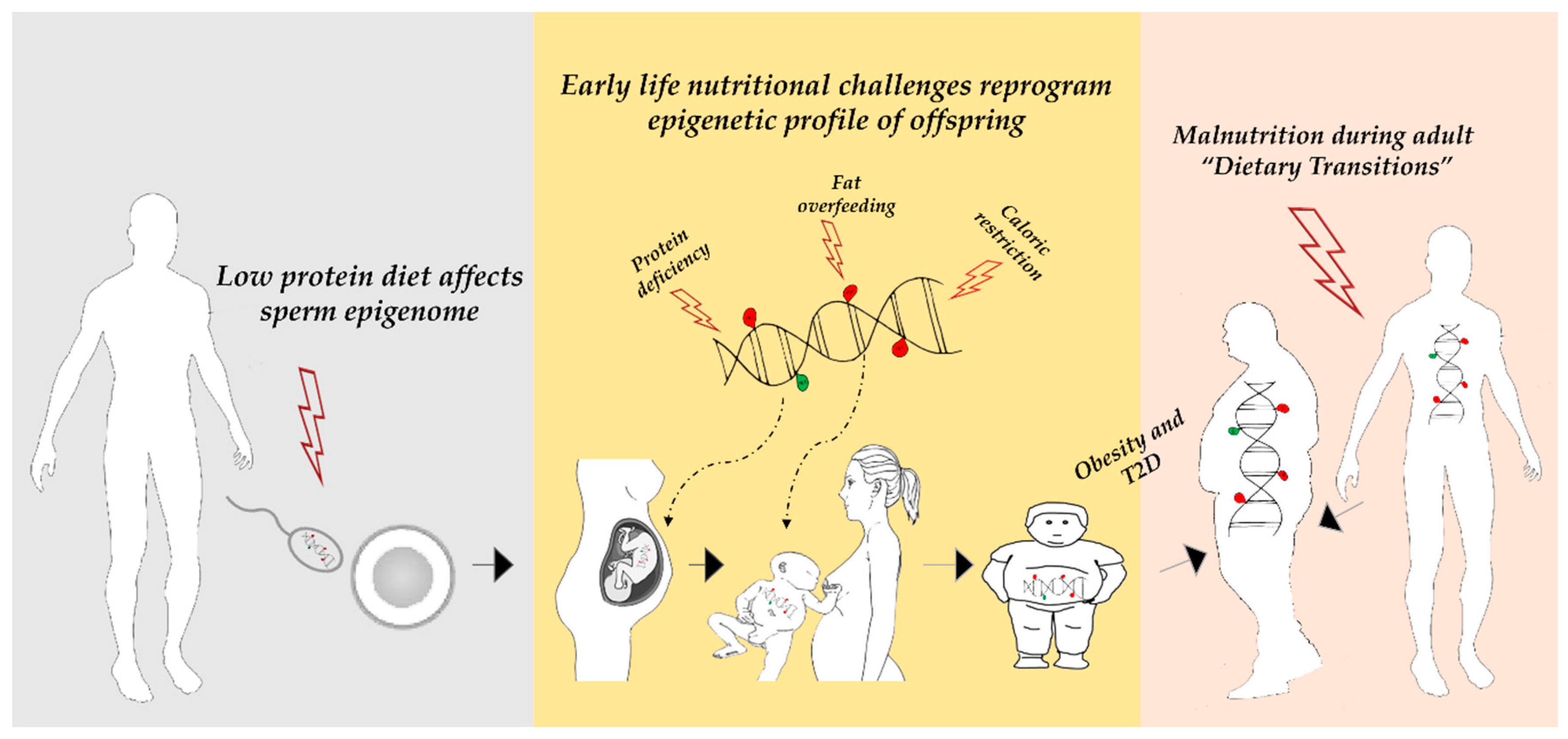
1. Introduction
2. DNA Methylation
3. Time Points of Plasticity in the Epigenetic System
4. The Agouti Mouse Model
5. Nutritional Factors Affecting DNA Methylation
5.1. Protein Malnutrition
5.2. Global Caloric Restriction
5.2.1. Animal Models
5.2.2. Human Studies
5.3. High-Fat Feeding
5.3.1. Animal Models
5.3.2. Human Studies
6. DNA Methylation and Future Medicine
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| T2D | Type 2 diabetes |
| GWAS | Genome-wide association studies |
| HFD | High-fat diet |
| DNMT | DNA methyltransferase |
| CpG | Cytidine-phosphate-Guanine |
| 5mC | 5-methyl cytosine |
| TET | Ten-eleven translocation |
| SAM | S.-adenosyl-methionine |
| CR | Caloric restriction |
| Avy | Agouti viable yellow |
| Agrp | Agouti related neuropeptide |
| IAP | Intracisternal A particle |
| GR | Glucocorticoid receptor |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| Lxrα | Liver X receptor alpha |
| Hnf4α | Hepatocyte nuclear factor 4 alpha |
| Nyp | Neuropeptide Y |
| Pomc | Pro-opiomelanocortin |
| LEP | Leptin |
| LPD | Low-protein diet |
| IGF2/H19 | Insulin like growth factor 2/ H19 imprinted maternally expressed transcript |
| PCK1 | phosphoenolpyruvate carboxykinase 1 |
| INSIGF2 | INS-IGF2 Readthrough |
| GNASAS1 | GNAS antisense RNA 1 |
| MEG3 | Maternally expressed 3 |
| IL-10 | Interleukin-10 |
| WT1 | Wilms tumor 1 |
| TNF-α | Tumor necrosis factor alpha |
| LBW | Low birth weight |
| NBW | Normal birth weight |
| PPARGC1A | Peroxisome proliferative activated receptor, gamma, coactivator 1 alpha |
| ADIPOQ | Adiponectin |
| Fgf21 | Fibroblast growth factor 21 |
| FA | Fatty acid |
| SCD1 | Stearoyl-coenzyme A desaturase 1 |
| WAT | White adipose tissue |
| ANKRD26 | Ankyrin repeat domain 26 |
| Hoxa5 | Homeobox A5 |
| 5’UTR | 5’ untranslated region |
| STD | Standard diet |
| RXRA | Retinoid X receptor alpha |
| FTO | FTO alpha-ketoglutarate dependent dioxygenase |
| IRX3 | Iroquois homeobox 3 |
| HIF3A | Hypoxia inducible factor 3 subunit alpha |
| Nnat | Neuronatin |
| Peg3 | Paternally expressed 3 |
| Trim-28 | Tripartite motif-containing 28 |
| ABCG1 | ATP binding cassette subfamily G member 1 |
| PHOSPHO1 | Phosphoethanolamine/phosphocholine phosphatase |
| SOCS3 | Suppressor of cytokine signaling 3 |
| SREBF1 | Sterol regulatory element binding transcription factor 1 |
| TXNIP | Thioredoxin interacting protein |
| BMI | Body mass index |
| HbA1c | Hemoglobin A1c |
| EWAS | Epigenome-wide association study |
| HDACi | Histone deacetylase inhibitors |
| VPA | Valproic acid |
| TSA | Trichostatin A |
| HAT | Histone acetyltransferases |
References
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Raciti, G.A.; Longo, M.; Parrillo, L.; Ciccarelli, M.; Mirra, P.; Ungaro, P.; Formisano, P.; Miele, C.; Béguinot, F. Understanding type 2 diabetes: From genetics to epigenetics. Acta Diabetol. 2015, 52, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zheng, L.; Almeida, F.A. Epigenetic reprogramming in metabolic disorders: Nutritional factors and beyond. J. Nutr. Biochem. 2018, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, A.; Spinelli, R.; Ciccarelli, M.; Nigro, C.; Miele, C.; Beguinot, F.; Raciti, G.A. Epigenetics: Spotlight on type 2 diabetes and obesity. J. Endocrinol. Invest. 2016, 39, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Multhaup, M.L.; Seldin, M.M.; Jaffe, A.E.; Lei, X.; Kirchner, H.; Mondal, P.; Li, Y.; Rodriguez, V.; Drong, A.; Hussain, M.; et al. Mouse-human experimental epigenetic analysis unmasks dietary targets and genetic liability for diabetic phenotypes. Cell Metab. 2015, 21, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Szabό, M.; Máte, B.; Csép, K.; Benedek, T. Epigenetic modification linked to T2D, the heritability gap, and potential targets. Biochem. Genet. 2018, 56, 553–574. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Almeida, F.A. Mitochondrial alteration in type 2 diabetes and obesity: An epigenetic link. Cell Cycle 2014, 13, 890–897. [Google Scholar] [CrossRef]
- Van Dijk, S.J.; Tellam, R.L.; Morrison, J.L.; Muhlhausler, B.S.; Molloy, P.L. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin. Epigenetics 2015, 7, 66. [Google Scholar] [CrossRef]
- Leung, A.; Parks, B.W.; Du, J.; Trac, C.; Setten, R.; Chen, Y.; Brown, K.; Lusis, A.J.; Natarajan, R.; Schones, D.E. Open chromatin profiling in mice livers reveals unique chromatin variations induced by high fat diet. J. Biol. Chem. 2014, 289, 23557–23567. [Google Scholar] [CrossRef]
- De Castro Barbosa, T.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjögren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2015, 5, 184–197. [Google Scholar] [CrossRef]
- Jacobsen, S.C.; Gillberg, L.; Bork-Jensen, J.; Ribel-Madsen, R.; Lara, E.; Calvanese, V.; Ling, C.; Fernandez, A.F.; Fraga, M.F.; Poulsen, P.; et al. Young men with low birthweight exhibit decreased plasticity of genome-wide muscle DNA methylation by high-fat overfeeding. Diabetologia 2014, 57, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Vickers, M.H. Early life nutrition, epigenetics and programming of later life disease. Nutrients 2014, 6, 2165–2178. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Epigenetic mechanism link maternal diets and gut microbiome to obesity in the offspring. Front Genet. 2018, 9, 342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kutateladze, T.G. Diet and the epigenome. Nat. Commun. 2018, 9, 3375. [Google Scholar] [CrossRef] [PubMed]
- Parrillo, L.; Costa, V.; Raciti, G.A.; Longo, M.; Spinelli, R.; Esposito, R.; Nigro, C.; Vastolo, V.; Desiderio, A.; Zatterale, F.; et al. Hoxa5 undergoes dynamic DNA methylation and transcriptional repression in the adipose tissue of mice exposed to high-fat diet. Int J. Obes. 2016, 40, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Raciti, G.A.; Nigro, C.; Longo, M.; Parrillo, L.; Miele, C.; Formisano, P.; Béguinot, F. Personalized medicine and type 2 diabetes: Lesson from epigenetics. Epigenomics 2014, 6, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.H.; Wang, H.T.; Kong, Q.P. Dynamic DNA methylation during aging: A “prophet” of age-related outcomes. Front Genet. 2019, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Jang, H.S.; Shin, W.J.; Lee, J.E.; Do, J.T. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes 2017, 8, 148. [Google Scholar] [CrossRef]
- Longo, M.; Raciti, G.A.; Zatterale, F.; Parrillo, L.; Desiderio, A.; Spinelli, R.; Hammarstedt, A.; Hedjazifar, S.; Hoffmann, J.M.; Nigro, C.; et al. Epigenetic modifications of the Zfp/ZNF423 gene control murine adipogenic commitment and are dysregulated in human hypertrophic obesity. Diabetologia 2018, 61, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef] [PubMed]
- Wolffe, A.P.; Jones, P.L.; Wade, P.A. DNA demethylation. Proc. Natl. Acad. Sci. USA 1999, 96, 5894–5896. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Lu, Y.; Jelinek, J.; Liang, S.; Estecio, M.R.; Barton, M.C.; Issa, J.P. TET1 is a maintenance DNA demethylase that prevents methylation spreading in differentiated cells. Nucleic Acids Res. 2014, 42, 6956–6971. [Google Scholar] [CrossRef]
- Ichiyama, K.; Chen, T.; Wang, X.; Yan, X.; Kim, B.S.; Tanaka, S.; Ndiaye-Lobry, D.; Deng, Y.; Zou, Y.; Zheng, P.; et al. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells. Immunity 2015, 42, 613–626. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef]
- Chao, M.P.; Seita, J.; Weissman, I.L. Establishment of a normal hematopoietic and leukemia stem cell hierarchy. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 439–449. [Google Scholar] [CrossRef]
- Farlik, M.; Halbritter, F.; Müller, F.; Choudry, F.A.; Ebert, P.; Klughammer, J.; Farrow, S.; Santoro, A.; Ciaurro, V.; Mathur, A.; et al. DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell 2016, 19, 808–822. [Google Scholar] [CrossRef]
- Côté, S.; Gagné-Ouellet, V.; Guay, S.P.; Allard, C.; Houde, A.A.; Perron, P.; Baillargeon, J.P.; Gaudet, D.; Guérin, R.; Brisson, D.; et al. PPARGC1α gene DNA methylation variations in human placenta mediate the link between maternal hyperglycemia and leptin levels in newborns. Clin. Epigenetics 2016, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.V.; Sheppard, B.; Windham, G.C.; Schieve, L.A.; Schendel, D.E.; Croen, L.A.; Chopra, P.; Alisch, R.S.; Newschaffer, C.J.; Warren, S.T.; et al. Case-control meta-analysis of blood DNA methylation and autism spectrum disorder. Mol. Autism. 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Rabinowitz, J.D. One-carbon metabolism in health and disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. Role of methionine on epigenetic modification of DNA methylation and gene expression in animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef]
- Langley-Evans, S.C. Nutrition in early life and the programming of adult disease: A review. J. Hum. Nutr. Diet. 2015, 28, 1–14. [Google Scholar] [CrossRef]
- Jiménez-Chillarón, J.C.; Díaz, R.; Martínez, D.; Pentinat, T.; Ramón-Krauel, M.; Ribó, S.; Plösch, T. The role of nutrition on epigenetic modifications and their implications on health. Biochimie 2012, 94, 2242–2263. [Google Scholar] [CrossRef]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Env. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef]
- Cropley, J.E.; Suter, C.M.; Beckman, K.B.; Martin, D.I. Germ-line epigenetic modification of the murine Avy allele by nutritional supplementation. Proc. Natl. Acad. Sci. USA 2006, 103, 17308–17312. [Google Scholar] [CrossRef]
- Rees, W.D.; Hay, S.M.; Brown, D.S.; Antipatis, C.; Palmer, R.M. Maternal protein deficiency causes hypermethylation of DNA in the livers of rat fetuses. J. Nutr. 2000, 130, 1821–1826. [Google Scholar] [CrossRef]
- Lillycrop, K.A.; Slater-Jefferies, J.L.; Hanson, M.A.; Godfrey, K.M.; Jackson, A.A.; Burdge, G.C. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br. J. Nutr. 2007, 97, 1064–1073. [Google Scholar]
- Lillycrop, K.A.; Phillips, E.S.; Torrens, C.; Hanson, M.A.; Jackson, A.A.; Burdge, G.C. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br. J. Nutr. 2008, 100, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Van Straten, E.M.; Bloks, V.W.; Huijkman, N.C.; Baller, J.F.; van Meer, H.; Lütjohann, D.; Kuipers, F.; Plösch, T. The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Sandovici, I.; Smith, N.H.; Nitert, M.D.; Ackers-Johnson, M.; Uribe-Lewis, S.; Ito, Y.; Jones, R.H.; Marquez, V.E.; Cairns, W.; Tadayyon, M.; et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proc. Natl. Acad. Sci. USA 2011, 108, 5449–5454. [Google Scholar] [CrossRef] [PubMed]
- Coupé, B.; Amarger, V.; Grit, I.; Benani, A.; Parnet, P. Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinology 2010, 151, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Jousse, C.; Parry, L.; Lambert-Langlais, S.; Maurin, A.C.; Averous, J.; Bruhat, A.; Carraro, V.; Tost, J.; Letteron, P.; Chen, P.; et al. Perinatal undernutrition affects the methylation and expression of the leptin gene in adults: Implication for the understanding of metabolic syndrome. FASEB J. 2011, 25, 3271–3278. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.J.; Dias, I.; Tsuro, H.; Allen, D.; Emes, R.D.; Moreton, J.; Wilson, R.; Ingram, R.J.M.; Sinclair, K.D. Paternal diet programs offspring health through sperm- and seminal plasma-specific pathways in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 10064–10069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Rattanatray, L.; MacLaughlin, S.M.; Cropley, J.E.; Suter, C.M.; Molloy, L.; Kleemann, D.; Walker, S.K.; Muhlhausler, B.S.; Morrison, J.L.; et al. Periconceptional undernutrition in normal and overweight ewes leads to increased adrenal growth and epigenetic changes in adrenal IGF2/H19 gene in offspring. FASEB J. 2010, 24, 2772–2782. [Google Scholar] [CrossRef] [PubMed]
- Perkins, E.; Murphy, S.K.; Murtha, A.P.; Schildkraut, J.; Jirtle, R.L.; Demark-Wahnefried, W.; Forman, M.R.; Kurtzberg, J.; Overcash, F.; Huang, Z.; et al. Insulin-like growth factor 2/H19 methylation at birth and risk of overweight and obesity in children. J. Pediatr. 2012, 161, 31–39. [Google Scholar] [CrossRef]
- Nijland, M.J.; Mitsuya, K.; Li, C.; Ford, S.; McDonald, T.J.; Nathanielsz, P.W.; Cox, L.A. Epigenetic modification of fetal baboon hepatic phosphoenolpyruvate carboxykinase following exposure to moderately reduced nutrient availability. J. Physiol. 2010, 588 Pt 8, 1349–1359. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Di Francesco, A.; Di Germanio, C.; Bernier, M.; de Cabo, R. A time to fast. Science 2018, 362, 770–775. [Google Scholar] [CrossRef]
- Campión, J.; Milagro, F.I.; Goyenechea, E.; Martínez, J.A. TNF-alpha promoter methylation as a predictive biomarker for weight-loss response. Obesity 2009, 17, 1293–1297. [Google Scholar]
- Milagro, F.I.; Mansego, M.L.; De Miguel, C.; Martínez, J.A. Dietary factors, epigenetic modifications and obesity outcomes: Progresses and perspectives. Mol. Asp. Med. 2013, 34, 782–812. [Google Scholar] [CrossRef]
- Bouchard, L.; Rabasa-Lhoret, R.; Faraj, M.; Lavoie, M.E.; Mill, J.; Pérusse, L.; Vohl, M.C. Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction. Am. J. Clin. Nutr. 2010, 91, 309–320. [Google Scholar] [CrossRef]
- Jørgensen, S.W.; Brøns, C.; Bluck, L.; Hjort, L.; Færch, K.; Thankamony, A.; Gillberg, L.; Friedrichsen, M.; Dunger, D.B.; Vaag, A.A. Metabolic response to 36 hours of fasting in young men born small vs. appropriate for gestational age. Diabetologia 2015, 58, 178–187. [Google Scholar] [CrossRef]
- Hjort, L.; Jørgensen, S.W.; Gillberg, L.; Hall, E.; Brøns, C.; Frystyk, J.; Vaag, A.A.; Ling, C. 36 h fasting of young men influences adipose tissue DNA methylation of LEP and ADIPOQ in a birth weight-dependent manner. Clin. Epigenetics 2017, 9, 40. [Google Scholar] [CrossRef]
- Samblas, M.; Milagro, F.I.; Martínez, A. DNA methylation markers in obesity, metabolic syndrome, and weight loss. Epigenetics 2019, 14, 421–444. [Google Scholar] [CrossRef]
- Vucetic, Z.; Kimmel, J.; Totoki, K.; Hollenbeck, E.; Reyes, T.M. Maternal high-fat diet alters methylation and gene expression of dopamine and opioid-related genes. Endocrinology 2010, 151, 4756–4764. [Google Scholar] [CrossRef]
- Ehara, T.; Kamei, Y.; Yuan, X.; Takahashi, M.; Kanai, S.; Tamura, E.; Tsujimoto, K.; Tamiya, T.; Nakagawa, Y.; Shimano, H.; et al. Ligand-activated PPARα-dependent DNA demethylation regulates the fatty acid β-oxidation genes in the postnatal liver. Diabetes 2015, 64, 775–784. [Google Scholar] [CrossRef]
- Yuan, X.; Tsujimoto, K.; Hashimoto, K.; Kawahori, K.; Hanzawa, N.; Hamaguchi, M.; Seki, T.; Nawa, M.; Ehara, T.; Kitamura, Y.; et al. Epigenetic modulation of Fgf21 in the perinatal mouse liver ameliorates diet-induced obesity in adulthood. Nat. Commun. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- BonDurant, L.D.; Ameka, M.; Naber, M.C.; Markan, K.R.; Idiga, S.O.; Acevedo, M.R.; Walsh, S.A.; Ornitz, D.M.; Potthoff, M.J. FGF21 Regulates metabolism through adipose-dependent and -independent mechanisms. Cell Metab. 2017, 25, 935.e4–944.e4. [Google Scholar] [CrossRef] [PubMed]
- Butruille, L.; Marousez, L.; Pourpe, C.; Oger, F.; Lecoutre, S.; Catheline, D.; Görs, S.; Metges, C.C.; Guinez, C.; Laborie, C.; et al. Maternal high-fat diet during suckling programs visceral adiposity and epigenetic regulation of adipose tissue stearoyl-CoA desaturase-1 in offspring. Int J. Obes. 2019. [Google Scholar] [CrossRef]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [PubMed]
- Raciti, G.A.; Spinelli, R.; Desiderio, A.; Longo, M.; Parrillo, L.; Nigro, C.; D’Esposito, V.; Mirra, P.; Fiory, F.; Pilone, V.; et al. Specific CpG hyper-methylation leads to Ankrd26 gene down-regulation in white adipose tissue of a mouse model of diet-induced obesity. Sci. Rep. 2017, 7, 43526. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, L.; Perfilyev, A.; Brøns, C.; Thomasen, M.; Grunnet, L.G.; Volkov, P.; Rosqvist, F.; Iggman, D.; Dahlman, I.; Risérus, U.; et al. Adipose tissue transcriptomics and epigenomics in low birthweight men and controls: Role of high-fat overfeeding. Diabetologia 2016, 59, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.C.; Brøns, C.; Bork-Jensen, J.; Ribel-Madsen, R.; Yang, B.; Lara, E.; Hall, E.; Calvanese, V.; Nilsson, E.; Jørgensen, S.W.; et al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia 2012, 55, 3341–3349. [Google Scholar] [CrossRef] [PubMed]
- Brøns, C.; Jacobsen, S.C.; Nilsson, E.; Ronn, T.; Jensen, C.B.; Storgaard, H.; Poulsen, P.; Groop, L.; Ling, C.; Asturp, A.; et al. Deoxyribonucleic acid methylation and gene expression of PPARGC1A in human muscle is influenced by high-fat overfeeding in a birth-weight-dependent manner. J. Clin. Endocrinol. Metab. 2010, 95, 3048–3056. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Willett, W.C.; Volek, J.S.; Neuhouser, M.L. Dietary fat: From foe to friend? Science 2018, 362, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Perfilyev, A.; Dahlman, I.; Gillberg, L.; Rosqvist, F.; Iggman, D.; Volkov, P.; Nilsson, E.; Risérus, U.; Ling, C. Impact of polyunsaturated and saturated fat overfeeding on the DNA-methylation pattern in human adipose tissue: A randomized controlled trial. Am. J. Clin. Nutr. 2017, 105, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Volkov, P.; Dayeh, T.; Bacos, K.; Rönn, T.; Nitert, M.D.; Ling, C. Effects of palmitate on genome-wide mRNA expression and DNA methylation patterns in human pancreatic islets. BMC Med. 2014, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Willmer, T.; Johnson, R.; Louw, J.; Pheiffer, C. Blood-based DNA methylation biomarkers for type 2 diabetes: Potential for clinical applications. Front. Endocrinol. 2018, 9, 744. [Google Scholar] [CrossRef] [PubMed]
- Oussalah, A.; Rischer, S.; Bensenane, M.; Conroy, G.; Filhine-Tresarrieu, P.; Debard, R.; Forest-Tramoy, D.; Josse, T.; Reinicke, D.; Garcia, M.; et al. Plasma mSEPT9: A novel circulating cell-free DNA-based epigenetic biomarker to diagnose hepatocellular carcinoma. EBioMedicine 2018, 30, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Gillberg, L.; Ling, C. The potential use of DNA methylation biomarkers to identify risk and progression of type 2 diabetes. Front. Endocrinol. 2015, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Toperoff, G.; Aran, D.; Kark, J.D.; Rosenberg, M.; Dubnikov, T.; Nissan, B.; Wainstein, J.; Friedlander, Y.; Levy-Lahad, E.; Glaser, B.; et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum. Mol. Genet. 2012, 21, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Smemo, S.; Tena, J.J.; Kim, K.H.; Gamazon, E.R.; Sakabe, N.J.; Gómez-Marín, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 20, 371–375. [Google Scholar] [CrossRef]
- Sharp, G.C.; Lawlor, D.A.; Richmond, R.C.; Fraser, A.; Simpkin, A.; Suderman, M.; Shihab, H.A.; Lyttleton, O.; McArdle, W.; Ring, S.M.; et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: Findings from the Avon longitudinal study of parents and children. Int. J. Epidemiol. 2015, 44, 1288–1304. [Google Scholar] [CrossRef]
- Dalgaard, K.; Landgraf, K.; Heyne, S.; Lempradl, A.; Longinotto, J.; Gossens, K.; Ruf, M.; Orthofer, M.; Strogantsev, R.; Selvaraj, M.; et al. Trim28 haploinsufficiency triggers bi-stable epigenetic obesity. Cell 2016, 164, 353–364. [Google Scholar] [CrossRef]
- Rhee, J.K.; Lee, J.H.; Yang, H.K.; Kim, T.M.; Yoon, K.H. DNA Methylation profiles of blood cells are distinct between early-onset obese and control individuals. Genom. Inf. 2017, 15, 28–37. [Google Scholar] [CrossRef]
- Dick, K.J.; Nelson, C.P.; Tsaprouni, L.; Sandling, J.K.; Aïssi, D.; Wahl, S.; Meduri, E.; Morange, P.E.; Gagnon, F.; Grallert, H.; et al. DNA methylation and body-mass index: A genome-wide analysis. Lancet 2014, 383, 1990–1998. [Google Scholar] [CrossRef]
- Chambers, J.C.; Loh, M.; Lehne, B.; Drong, A.; Kriebel, J.; Motta, V.; Wahl, S.; Elliott, H.R.; Rota, F.; Scott, W.R.; et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: A nested case-control study. Lancet Diabetes Endocrinol. 2015, 3, 526–534. [Google Scholar] [CrossRef]
- Dayeh, T.; Tuomi, T.; Almgren, P.; Perfilyev, A.; Jansson, P.A.; de Mello, V.D.; Pihlajamäki, J.; Vaag, A.; Groop, L.; Nilsson, E.; et al. DNA methylation of loci within ABCG1 and PHOSPHO1 in blood DNA is associated with future type 2 diabetes risk. Epigenetics 2016, 11, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Kriebel, J.; Herder, C.; Rathmann, W.; Wahl, S.; Kunze, S.; Molnos, S.; Volkova, N.; Schramm, K.; Carstensen-Kirberg, M.; Waldenberger, M.; et al. Association between DNA methylation in whole blood and measures of glucose metabolism: KORA F4 study. PLoS ONE 2016, 11, e0152314. [Google Scholar] [CrossRef] [PubMed]
- Walaszczyk, E.; Luijten, M.; Spijkerman, A.M.W.; Bonder, M.J.; Lutgers, H.L.; Snieder, H.; Wolffenbuttel, B.H.R.; van Vliet-Ostaptchouk, J.V. DNA methylation markers associated with type 2 diabetes, fasting glucose and HbA1c levels: A systematic review and replication in a case-control sample of the lifelines study. Diabetologia 2018, 61, 354–368. [Google Scholar] [CrossRef]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arter. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef]
- Bacos, K.; Gillberg, L.; Volkov, P.; Olsson, A.H.; Hansen, T.; Pedersen, O.; Gjesing, A.P.; Eiberg, H.; Tuomi, T.; Almgren, P.; et al. Blood-based biomarkers of age-associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat. Commun. 2016, 7, 11089. [Google Scholar] [CrossRef]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.-C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef]
- Cordero, P.; Campion, J.; Milagro, F.I.; Goyenechea, E.; Steemburgo, T.; Javierre, B.M.; Martinez, J.A. Leptin and TNF-α promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J. Physiol. Biochem. 2011, 67, 463–470. [Google Scholar] [CrossRef]
- Huang, Y.T.; Maccani, J.Z.J.; Hawley, N.L.; Wing, R.R.; Kelsey, K.T.; McCaffery, J.M. Epigenetic patterns in successful weight loss maintainers: A pilot study. Int J. Obes. 2015, 39, 865–868. [Google Scholar] [CrossRef]
- Nicoletti, C.F.; Nonino, C.B.; de Oliveira, B.A.; Pinhel, M.A.; Mansego, M.L.; Milagro, F.I.; Zulet, M.A.; Martinez, J.A. DNA methylation and hydroxymethylation levels in relation to two weight loss strategies: Energy-restricted diet or bariatric surgery. Obes. Surg. 2016, 26, 603–611. [Google Scholar] [CrossRef] [PubMed]
- McEwen, L.M.; Gatev, E.G.; Jones, M.J.; MacIsaac, J.L.; McAllister, M.M.; Goulding, R.E.; Madden, K.M.; Dawes, M.G.; Kobor, M.S.; Ashe, M.C. DNA methylation signatures in peripheral blood mononuclear cells from a lifestyle intervention for women at midlife: A pilot randomized controlled trial. Appl. Physiol. Nutr. Metab. 2018, 43, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Hotte, S.J.; Chen, E.X.; Hirte, H.W.; Oza, A.M.; Moretto, P.; Webster, S.; Laughlin, A.; Stayner, L.A.; McGill, S.; et al. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin. Cancer Res. 2009, 27, 3528. [Google Scholar] [CrossRef] [PubMed]
- Lundh, M.; Galbo, T.; Poulsen, S.S.; Mandrup-Poulsen, T. Histone deacetylase 3 inhibition improves glycaemia and insulin secretion in obese diabetic rats. Diabetes Obes. Metab. 2015, 17, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Daneshpajooh, M.; Eliasson, L.; Bacos, K.; Ling, C. MC1568 improves insulin secretion in islets from type 2 diabetes patients and rescues β-cell dysfunction caused by Hdac7 upregulation. Acta Diabetol. 2018, 55, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Daneshpajooh, M.; Bacos, K.; Bysani, M.; Bagge, A.; Ottosson Laakso, E.; Vikman, P.; Eliasson, L.; Mulder, H.; Ling, C. HDAC7 is overexpressed in human diabetic islets and impairs insulin secretion in rat islets and clonal beta cells. Diabetologia 2017, 60, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Zuo, S.; Fries, B.E.; Szafara, K.; Regal, R. Valproic Acid as a potentiator of metabolic syndrome in institutionalized residents on concomitant antipsychotics: Fat chance, or slim to none? Pharm. Ther. 2015, 40, 126–132. [Google Scholar]
- Avery, L.B.; Bumpus, N.N. Valproic acid is a novel activator of AMP-activated protein kinase and decreases liver mass, hepatic fat accumulation, and serum glucose in obese mice. Mol. Pharm. 2014, 85, 1–10. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef]
- Timp, W.; Feinberg, A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer. 2013, 13, 497–510. [Google Scholar] [CrossRef]
- Kirchner, H.; Sinha, I.; Gao, H.; Ruby, M.A.; Schönke, M.; Lindvall, J.M.; Barrès, R.; Krook, A.; Näslund, E.; Dahlman-Wright, K.; et al. Altered DNA methylation of glycolytic and lipogenic genes in liver from obese and type 2 diabetic patients. Mol. Metab. 2016, 5, 171–183. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Dietary Condition | DNA Methylation Regulated Locus(i) | Sample Type(s) | Species | References |
|---|---|---|---|---|
| Low protein diet | GR, PPARα, Hnf4a, Nyp, Pomc | Liver, pancreatic islets, hypothalamus | Rat | [40,41,43,44] |
| Lxrα, Lep | Liver, adipose tissue | Mouse | [42,45] | |
| Caloric restriction | IGF2/H19 | Adrenal gland | Sheep | [47] |
| PCK1 | Fetal liver | Baboon | [49] | |
| IGF2, INSIGF2, GNASAS1, MEG3, IL-10, LEP | Blood | Human | [50,51] | |
| WT1, TNF-α | Blood | Human | [53,54] | |
| PPARGC1A | Skeletal muscle | Human | [56] | |
| ADIPOQ, LEP | Adipose tissue | Human | [57] | |
| High-fat feeding | Fgf21 | Liver | Mouse | [61] |
| Scd1 | Adipose tissue | Rat | [63] | |
| Ankrd26, Hoxa5 | Adipose tissue | Mouse | [15,65] |
| Study Population | Sample Type | Methylation Strategy | Epigenetically Regulated Genes | Reference |
|---|---|---|---|---|
| Healthy men (n = 21) | Skeletal muscle | Genome-wide * | DNM2, MGMT, SLC2A3/GLUT3, MRC1, ACAT2, APOH, DCC, ESRRG, FOLH1, GTF2I, MC4R, MYST4, AKT2, PDX1/IPF1, SLC30A8, CDKN2A, CDKN2B, PPARG | [67] |
| NBW (n = 23), LBW (n = 17) | Skeletal muscle | Genome-wide * | IGF2R, TNF, CDKN2B, KCNJ11, KCNQ1, GABRA3, UGT2B7, FOLH1, FUT1, NDUFS2, FAP | [11] |
| NBW (n = 24), LBW (n = 16) | Subcutaneous adipose tissue | Genome-wide * | ACAT1, CPLX1, FADS2, GPRC5B, HCCA2, IGF2R, CIDEA, KLF14, PRDM16 | [66] |
| NBW (n = 26), LBW (n = 20) | Skeletal muscle | Gene-specific | PPARGC1A | [68] |
| Study Population | Sample Type | Methylation Strategy | Epigenetically Regulated Genes | Reference |
|---|---|---|---|---|
| LIPOGAIN cohort (n = 31; SFA group n = 17; PUFA group n = 14) | Subcutaneous adipose tissue | Genome-wide * | RPSAP9, ADIPOQ, FABP1, FABP2, FABP7, MC2R, XKR4, MC3R, MC5R, PPARGC1A, TNF, ACO1, SLC37A2, AR, CXCL2, FOXO1, FTO, IL6, INSR, MC1R, MC3R, MEF2A, NEGR1, POMC | [70] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrillo, L.; Spinelli, R.; Nicolò, A.; Longo, M.; Mirra, P.; Raciti, G.A.; Miele, C.; Beguinot, F. Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges. Int. J. Mol. Sci. 2019, 20, 2983. https://doi.org/10.3390/ijms20122983
Parrillo L, Spinelli R, Nicolò A, Longo M, Mirra P, Raciti GA, Miele C, Beguinot F. Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges. International Journal of Molecular Sciences. 2019; 20(12):2983. https://doi.org/10.3390/ijms20122983
Chicago/Turabian StyleParrillo, Luca, Rosa Spinelli, Antonella Nicolò, Michele Longo, Paola Mirra, Gregory Alexander Raciti, Claudia Miele, and Francesco Beguinot. 2019. "Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges" International Journal of Molecular Sciences 20, no. 12: 2983. https://doi.org/10.3390/ijms20122983
APA StyleParrillo, L., Spinelli, R., Nicolò, A., Longo, M., Mirra, P., Raciti, G. A., Miele, C., & Beguinot, F. (2019). Nutritional Factors, DNA Methylation, and Risk of Type 2 Diabetes and Obesity: Perspectives and Challenges. International Journal of Molecular Sciences, 20(12), 2983. https://doi.org/10.3390/ijms20122983