New Small Molecule Drugs for Thrombocytopenia: Chemical, Pharmacological, and Therapeutic Use Considerations



Abstract
:1. Introduction
2. The Development of Small Molecule Drugs for Thrombocytopenia
2.1. Thrombopoietin Receptor (TPO-R)
2.2. Spleen Tyrosine Kinase (SYK)
3. The New Approvals
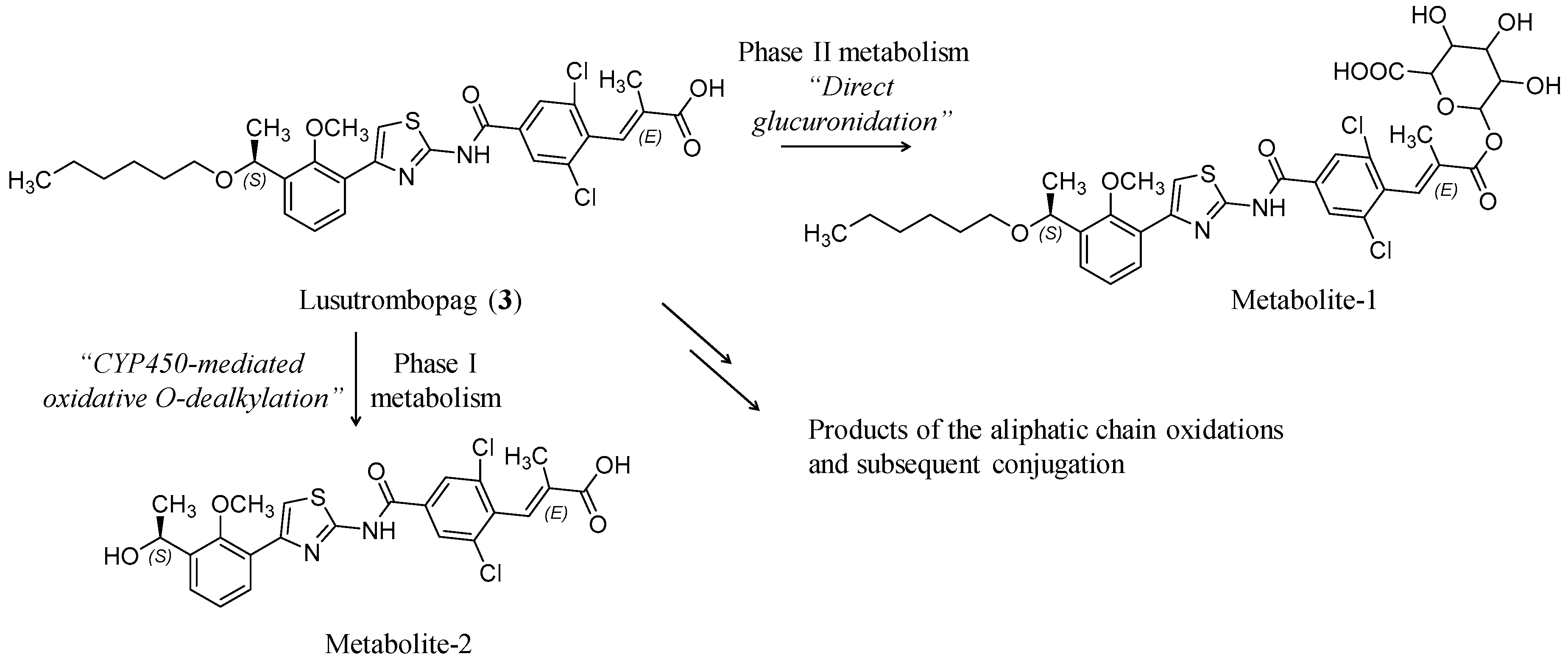
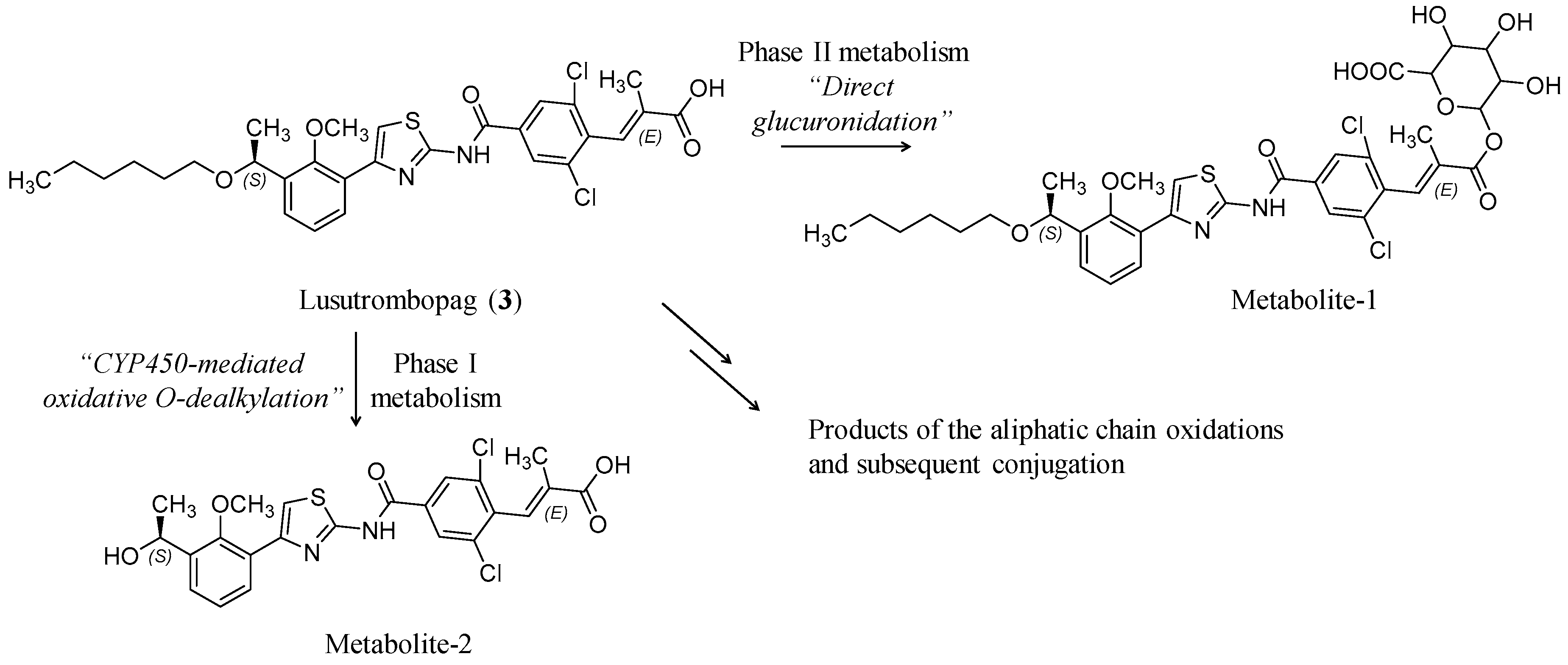
3.1. Lusutrombopag
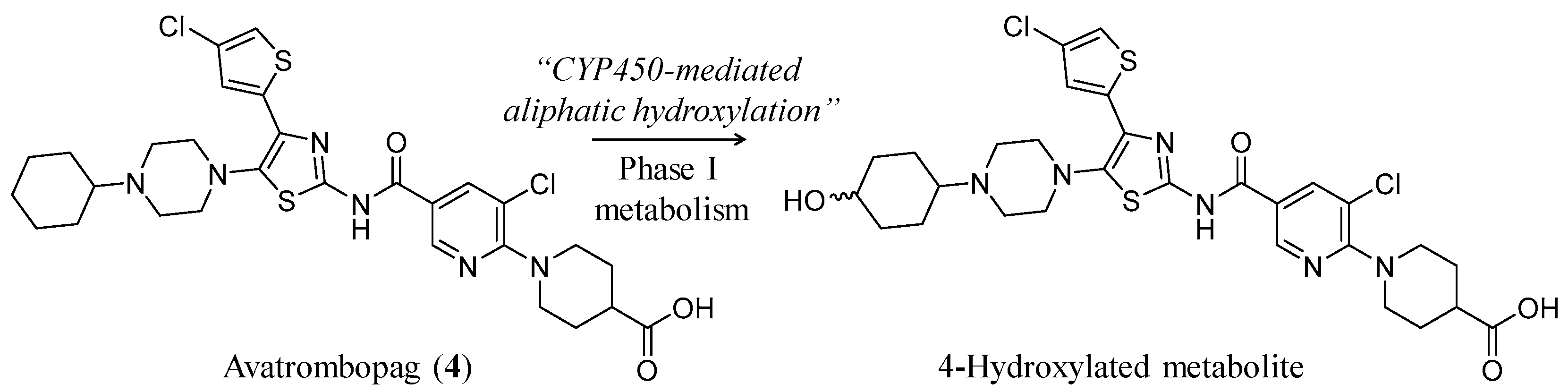
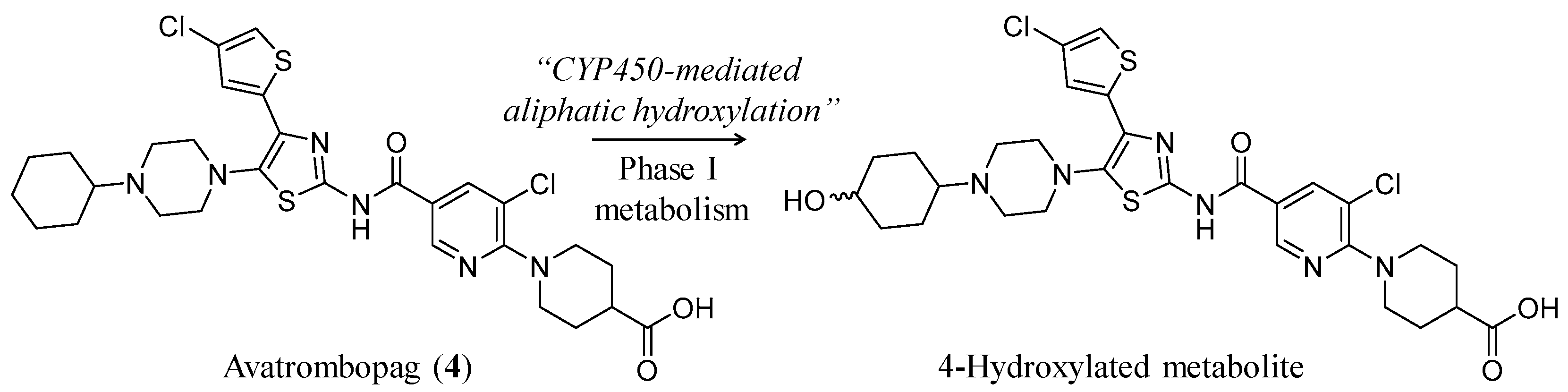
3.2. Avatrombopag
3.3. Fostamatinib
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCRP | Breast cancer resistance protein |
| HIT | Heparin-induced thrombocytopenia |
| JAK | Janus kinase |
| MAPK | Mitogen-activated protein kinase |
| PI3K | Phosphatidylinositol-3 kinase |
| STAT | Signal transducer and activator of transcription |
| SYK | Spleen tyrosine kinase |
| TPO-R | Thrombopoietin receptor |
References
- Izak, M.; Bussel, J.B. Management of thrombocytopenia. F1000Prime Rep. 2014, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Cines, D.B.; Levine, L.D. Thrombocytopenia in pregnancy. Blood 2017, 130, 2271–2277. [Google Scholar] [CrossRef] [Green Version]
- Gafter-Gvili, A.; Mansur, N.; Bivas, A.; Zemer-Wassercug, N.; Bishara, J.; Leibovici, L.; Paul, M. Thrombocytopenia in Staphylococcus aureus bacteremia: Risk factors and prognostic importance. Mayo Clin. Proc. 2011, 86, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Aster, R.H.; Bougie, D.W. Drug-induced immune thrombocytopenia. N. Engl. J. Med. 2007, 357, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Aster, R.H.; Curtis, B.R.; McFarland, J.G.; Bougie, D.W. Drug-induced immune thrombocytopenia: Pathogenesis, diagnosis and management. J. Thromb. Haemost. 2009, 7, 911–918. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Wenham, T. Heparin-induced thrombocytopenia. Postgrad. Med. J. 2018, 94, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Arepally, G.M. Heparin-induced thrombocytopenia. Blood 2017, 129, 2864–2872. [Google Scholar] [CrossRef]
- Cooper, N. State of the art—How I manage immune thrombocytopenia. Br. J. Haematol. 2017, 177, 39–54. [Google Scholar] [CrossRef]
- Kado, R.; McCune, W.J. Treatment of primary and secondary immune thrombocytopenia. Curr. Opin. Rheumatol. 2019, 31, 213–222. [Google Scholar] [CrossRef]
- Kuter, D.J. Biology and chemistry of thrombopoietic agents. Semin. Hematol. 2010, 47, 243–248. [Google Scholar] [CrossRef]
- Varghese, L.N.; Defour, J.P.; Pecquet, C.; Constantinescu, S.N. The thrombopoietin receptor: Structural basis of traffic and activation by ligand, mutations, agonists, and mutated calreticulin. Front. Endocrinol. (Lausanne) 2017, 8, 59. [Google Scholar] [CrossRef]
- Kaushansky, K. Lineage-specific hematopoietic growth factors. N. Engl. J. Med. 2006, 354, 2034–2045. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, K.; Ishii, E.; Tsuji, K.; Yamamoto, S.; Yamaguchi, H.; Hara, T.; Koga, H.; Nakahata, T.; Miyazaki, S. Defective response to thrombopoietin and impaired expression of c-mpl mRNA of bone marrow cells in congenital amegakaryocytic thrombocytopenia. Br. J. Haematol. 1997, 96, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Ballmaier, M.; Germeshausen, M.; Schulze, H.; Cherkaoui, K.; Lang, S.; Gaudig, A.; Krukemeier, S.; Eilers, M.; Strauss, G.; Welte, K. C-mpl mutations are the cause of congenital amegakaryocytic thrombocytopenia. Blood 2001, 97, 139–146. [Google Scholar] [CrossRef]
- Ihara, K.; Ishii, E.; Eguchi, M.; Takada, H.; Suminoe, A.; Good, R.A.; Hara, T. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proc. Natl. Acad. Sci. USA 1999, 96, 3132–3136. [Google Scholar] [CrossRef]
- Stasi, R.; Bosworth, J.; Rhodes, E.; Shannon, M.S.; Willis, F.; Gordon-Smith, E.C. Thrombopoietic agents. Blood Rev. 2010, 24, 179–190. [Google Scholar] [CrossRef]
- Imbach, P.; Crowther, M. Thrombopoietin-receptor agonists for primary immune thrombocytopenia. N. Engl. J. Med. 2011, 365, 734–741. [Google Scholar] [CrossRef]
- Maan, R.; de Knegt, R.J.; Veldt, B.J. Management of thrombocytopenia in chronic liver disease: Focus on pharmacotherapeutic strategies. Drugs 2015, 75, 1981–1992. [Google Scholar] [CrossRef]
- Mitchell, W.B.; Bussel, J.B. Thrombopoietin receptor agonists: A critical review. Semin. Hematol. 2015, 52, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Cwirla, S.E.; Balasubramanian, P.; Duffin, D.J.; Wagstrom, C.R.; Gates, C.M.; Singer, S.C.; Davis, A.M.; Tansik, R.L.; Mattheakis, L.C.; Boytos, C.M.; et al. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science 1997, 276, 1696–1699. [Google Scholar] [CrossRef] [PubMed]
- Broudy, V.C.; Lin, N.L. AMG531 stimulates megakaryopoiesis in vitro by binding to Mpl. Cytokine 2004, 25, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nichol, J.L.; Sullivan, J.T. Pharmacodynamics and pharmacokinetics of AMG 531, a novel thrombopoietin receptor ligand. Clin. Pharm Therapeut. 2004, 76, 628–638. [Google Scholar] [CrossRef]
- Kuter, D.J.; Bussel, J.B.; Lyons, R.M.; Pullarkat, V.; Gernsheimer, T.B.; Senecal, F.M.; Aledort, L.M.; George, J.N.; Kessler, C.M.; Sanz, M.A.; et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: A double-blind randomised controlled trial. Lancet 2008, 371, 395–403. [Google Scholar] [CrossRef]
- Duffy, K.J.; Darcy, M.G.; Delorme, E.; Dillon, S.B.; Eppley, D.F.; Erickson-Miller, C.; Giampa, L.; Hopson, C.B.; Huang, Y.; Keenan, R.M.; et al. Hydrazinonaphthalene and azonaphthalene thrombopoietin mimics are nonpeptidyl promoters of megakaryocytopoiesis. J. Med. Chem. 2001, 44, 3730–3745. [Google Scholar] [CrossRef] [PubMed]
- Erickson-Miller, C.L.; DeLorme, E.; Tian, S.S.; Hopson, C.B.; Stark, K.; Giampa, L.; Valoret, E.I.; Duffy, K.J.; Luengo, J.L.; Rosen, J.; et al. Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp. Hematol. 2005, 33, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Erickson-Miller, C.L.; Delorme, E.; Tian, S.S.; Hopson, C.B.; Landis, A.J.; Valoret, E.I.; Sellers, T.S.; Rosen, J.; Miller, S.G.; Luengo, J.I.; et al. Preclinical activity of eltrombopag (SB-497115), an oral, nonpeptide thrombopoietin receptor agonist. Stem Cells. 2009, 27, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Yamane, N.; Tanaka, Y.; Ohyabu, N.; Yamane, S.; Maekawa, K.; Ishizaki, J.; Suzuki, R.; Itoh, T.; Takemoto, H. Characterization of novel non-peptide thrombopoietin mimetics, their species specificity and the activation mechanism of the thrombopoietin receptor. Eur. J. Pharmacol. 2008, 586, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Nakamura, S.; Taniguchi, T.; Yamamura, H. Purification and characterization of a cytosolic protein-tyrosine kinase from porcine spleen. Eur. J. Biochem. 1990, 188, 535–540. [Google Scholar] [CrossRef]
- Liu, D.; Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J. Hematol. Oncol. 2017, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Masuda, E.S.; Payan, D.G. Discovery and development of spleen tyrosine kinase (SYK) inhibitors. J. Med. Chem. 2012, 55, 3614–3643. [Google Scholar] [CrossRef]
- Takayama, M.; Yamada, H.; Takemoto, H.; Shiota, T.; Tanaka, Y.; Yamane, N.; Takahashi, K.; Oyabu, N.; Kuwabara, K.; Oshima, I.; et al. Discovery and Biological Evaluation of Lusutrombopag (S-888711) as a Novel Nonpeptide Drug Candidate for Thrombocytopenia. In Proceedings of the 247th ACS National Meeting & Exposition Abstracts of Papers, Dallas, TX, USA, 16–20 March 2014. MEDI-101. [Google Scholar]
- Yoshida, H.; Yamada, H.; Nogami, W.; Dohi, K.; Kurino-Yamada, T.; Sugiyama, K.; Takahashi, K.; Gahara, Y.; Kitaura, M.; Hasegawa, M.; et al. Development of a new knock-in mouse model and evaluation of pharmacological activities of lusutrombopag, a novel, nonpeptidyl small-molecule agonist of the human thrombopoietin receptor c-Mpl. Exp. Hematol. 2018, 59, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S. Lusutrombopag: First global approval. Drugs 2016, 76, 155–158. [Google Scholar] [CrossRef] [PubMed]
- FDA Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210923s000lbl.pdf (accessed on 14 April 2019).
- Hidaka, H.; Kurosaki, M.; Tanaka, H.; Kudo, M.; Abiru, S.; Igura, T.; Ishikawa, T.; Seike, M.; Katsube, T.; Ochiai, T.; et al. Lusutrombopag reduces need for platelet transfusion in patients with thrombocytopenia undergoing invasive procedures. Clin. Gastroenterol. Hepatol. 2019, 17, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, R.; Seike, M.; Kudo, M.; Tamai, H.; Kawazoe, S.; Katsube, T.; Ochiai, T.; Fukuhara, T.; Kano, T.; Tanaka, K.; et al. A randomized controlled trial of lusutrombopag in Japanese patients with chronic liver disease undergoing radiofrequency ablation. J. Gastroenterol. 2019, 54, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Katsube, T.; Ishibashi, T.; Kano, T.; Wajima, T. Population pharmacokinetic and pharmacodynamic modeling of lusutrombopag, a newly developed oral thrombopoietin receptor agonist, in healthy subjects. Clin. Pharmacokinet. 2016, 55, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Izumi, N.; Osaki, Y.; Yamamoto, K.; Kuro-kawa, M.; Tanaka, K.; Kano, T.; Fukuhara, T.; Ochiai, T.; Imawari, M. A phase 3, randomized, double-blind, placebo-controlled study of lusutrombopag for thrombocytopenia in patients with chronic liver disease undergoing elective invasive procedures in Japan (L-PLUS 1). Hepatology 2015, 62, 1397A–1398A. [Google Scholar]
- Peck-Radosavljevic, M.; Simon, K.; Iacobellis, A.; Hassanein, T.; Kayali, Z.; Tran, A.; Makara, M.; Ben Ari, Z.; Braun, M.; Mitrut, P.; et al. Lusutrombopag for the treatment of thrombocytopenia in patients with chronic liver disease undergoing invasive procedures (L-PLUS 2). Hepatology 2019. [Google Scholar] [CrossRef]
- Ishikawa, T.; Okoshi, M.; Tomiyoshi, K.; Kojima, Y.; Horigome, R.; Imai, M.; Nozawa, Y.; Iwanaga, A.; Sano, T.; Honma, T.; et al. Efficacy and safety of repeated use of lusutrombopag prior to radiofrequency ablation in patients with recurrent hepatocellular carcinoma and thrombocytopenia. Hepatol. Res. 2019. [Google Scholar] [CrossRef]
- Kotani, S.; Kohge, N.; Tsukano, K.; Ogawa, S.; Yamanouchi, S.; Kusunoki, R.; Aimi, M.; Miyaoka, Y.; Fujishiro, H. Avoidance of platelet transfusion with readministration of lusutrombopag before radiofrequency ablation in hepatocellular carcinoma: A case report. Nihon Shokakibyo Gakkai Zasshi 2017, 114, 1853–1859. [Google Scholar] [CrossRef]
- Sato, S.; Miyake, T.; Kataoka, M.; Isoda, K.; Yazaki, T.; Tobita, H.; Ishimura, N.; Kinoshita, Y. Efficacy of repeated lusutrombopag administration for thrombocytopenia in a patient scheduled for invasive hepatocellular carcinoma treatment. Intern. Med. 2017, 56, 2887–2890. [Google Scholar] [CrossRef]
- Sakamaki, A.; Watanabe, T.; Abe, S.; Kamimura, K.; Tsuchiya, A.; Takamura, M.; Kawai, H.; Yamagiwa, S.; Terai, S. Lusutrombopag increases hematocytes in a compensated liver cirrhosis patient. Clin. J. Gastroenterol. 2017, 10, 261–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, M.; Abe, K.; Hayashi, M.; Okai, K.; Takahashi, A.; Ohira, H. Two cases of liver cirrhosis treated with lusutrombopag before partial splenic embolization. Fukushima J. Med. Sci. 2017, 63, 165–171. [Google Scholar] [CrossRef]
- Takada, H.; Kurosaki, M.; Nakanishi, H.; Takahashi, Y.; Itakura, J.; Tsuchiya, K.; Yasui, Y.; Tamaki, N.; Takaura, K.; Komiyama, Y.; et al. Real-life experience of lusutrombopag for cirrhotic patients with low platelet counts being prepared for invasive procedures. PLoS ONE 2019, 14, e0211122. [Google Scholar] [CrossRef] [PubMed]
- Katano, T.; Sanada, Y.; Okada, N.; Mizuta, K. Lusutrombopag as pretreatment for liver biopsy following liver transplantation. Pediatr. Int. 2018, 60, 1033–1034. [Google Scholar] [CrossRef] [PubMed]
- Uojima, H.; Arase, Y.; Itokawa, N.; Atsukawa, M.; Satoh, T.; Miyazaki, K.; Hidaka, H.; Sung, J.H.; Kako, M.; Tsuruya, K.; et al. Relationship between response to lusutrombopag and splenic volume. World J. Gastroenterol. 2018, 24, 5271–5279. [Google Scholar] [CrossRef]
- Fukushima-Shintani, M.; Suzuki, K.; Iwatsuki, Y.; Abe, M.; Sugasawa, K.; Hirayama, F.; Kawasaki, T.; Nakahata, T. AKR-501 (YM477) a novel orally-active thrombopoietin receptor agonist. Eur. J. Haematol. 2009, 82, 247–254. [Google Scholar] [CrossRef]
- FDA Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210238s000lbl.pdf (accessed on 16 April 2019).
- Shirley, M. Avatrombopag: First global approval. Drugs 2018, 78, 1163–1168. [Google Scholar] [CrossRef]
- Długosz-Danecka, M.; Zdziarska, J.; Jurczak, W. Avatrombopag for the treatment of immune thrombocytopenia. Expert Rev. Clin. Immunol. 2019, 15, 327–339. [Google Scholar] [CrossRef]
- Nomoto, M.; Pastino, G.; Rege, B.; Aluri, J.; Ferry, J.; Han, D. Pharmacokinetics, pharmacodynamics, pharmacogenomics, safety, and tolerability of avatrombopag in healthy Japanese and white subjects. Clin. Pharmacol. Drug Dev. 2018, 7, 188–195. [Google Scholar] [CrossRef]
- Fukushima-Shintani, M.; Suzuki, K.; Iwatsuki, Y.; Abe, M.; Sugasawa, K.; Hirayama, F.; Kawasaki, T. AKR-501 (YM477) in combination with thrombopoietin enhances human megakaryocytopoiesis. Exp. Hematol. 2008, 36, 1337–1342. [Google Scholar] [CrossRef]
- Michelson, A.D.; Smolensky Koganov, E.; Forde, E.E.; Carmichael, S.L.; Frelinger, A.L., 3rd. Avatrombopag increases platelet count but not platelet activation in patients with thrombocytopenia resulting from liver disease. J. Thromb. Haemost. 2018, 16, 2515–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Samkari, H. Avatrombopag maleate for the treatment of periprocedural thrombocytopenia in patients with chronic liver disease. Drugs Today (Barc) 2018, 54, 647–655. [Google Scholar] [CrossRef]
- Nomoto, M.; Ferry, J.; Hussein, Z. Population pharmacokinetic/pharmacodynamic analyses of avatrombopag in patients with chronic liver disease and optimal dose adjustment guide with concomitantly administered CYP3A and CYP2C9 inhibitors. J. Clin. Pharmacol. 2018, 58, 1629–1638. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, M.; Zamora, C.A.; Schuck, E.; Boyd, P.; Chang, M.K.; Aluri, J.; Siu, Y.A.; Lai, W.G.; Yasuda, S.; Ferry, J.; et al. Pharmacokinetic/pharmacodynamic drug-drug interactions of avatrombopag when coadministered with dual or selective CYP2C9 and CYP3A interacting drugs. Br. J. Clin. Pharmacol. 2018, 84, 952–960. [Google Scholar] [CrossRef] [Green Version]
- Terrault, N.; Chen, Y.C.; Izumi, N.; Kayali, Z.; Mitrut, P.; Tak, W.Y.; Allen, L.F.; Hassanein, T. Avatrombopag before procedures reduces need for platelet transfusion in patients with chronic liver disease and thrombocytopenia. Gastroenterology 2018, 155, 705–718. [Google Scholar] [CrossRef]
- Markham, A. Fostamatinib: First global approval. Drugs 2018, 78, 959–963. [Google Scholar] [CrossRef] [PubMed]
- FDA Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/209299lbl.pdf (accessed on 17 April 2019).
- Sweeny, D.J.; Li, W.; Clough, J.; Bhamidipati, S.; Singh, R.; Park, G.; Baluom, M.; Grossbard, E.; Lau, D.T. Metabolism of fostamatinib, the oral methylene phosphate prodrug of the spleen tyrosine kinase inhibitor R406 in humans: Contribution of hepatic and gut bacterial processes to the overall biotransformation. Drug Metab. Dispos. 2010, 38, 1166–1176. [Google Scholar] [CrossRef]
- Rolf, M.G.; Curwen, J.O.; Veldman-Jones, M.; Eberlein, C.; Wang, J.; Harmer, A.; Hellawell, C.J.; Braddock, M. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol. Res. Perspect. 2015, 3, e00175. [Google Scholar] [CrossRef]
- Braselmann, S.; Taylor, V.; Zhao, H.; Wang, S.; Sylvain, C.; Baluom, M.; Qu, K.; Herlaar, E.; Lau, A.; Young, C.; et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J. Pharmacol. Exp. Ther. 2006, 319, 998–1008. [Google Scholar] [CrossRef]
- Podolanczuk, A.; Lazarus, A.H.; Crow, A.R.; Grossbard, E.; Bussel, J.B. Of mice and men: An open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 2009, 113, 3154–3160. [Google Scholar] [CrossRef]
- Bussel, J.; Arnold, D.M.; Grossbard, E.; Mayer, J.; Treliński, J.; Homenda, W.; Hellmann, A.; Windyga, J.; Sivcheva, L.; Khalafallah, A.A.; et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: Results of two phase 3, randomized, placebo-controlled trials. Am. J. Hematol. 2018, 93, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, T.; Martin, P.; Gillen, M.; Mathews, D.; Lisbon, E.; Kruusmägi, M. Effects of ranitidine (antacid), food, and formulation on the pharmacokinetics of fostamatinib: Results from five phase I clinical studies. Eur. J. Clin. Pharmacol. 2017, 73, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Lengel, D.; Lamm Bergström, E.; Barthlow, H.; Oldman, K.; Musgrove, H.; Harmer, A.; Valentin, J.P.; Duffy, P.; Braddock, M.; Curwen, J. Prevention of fostamatinib-induced blood pressure elevation by antihypertensive agents. Pharmacol. Res. Perspect. 2015, 3, e00176. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Gillen, M.; Millson, D.; Oliver, S.; Brealey, C.; Grossbard, E.B.; Baluom, M.; Lau, D.; Sweeny, D.; Mant, T.; et al. Effects of CYP3A4 inhibitors ketoconazole and verapamil and the CYP3A4 inducer Rifampicin on the pharmacokinetic parameters of fostamatinib: Results from in vitro and phase I clinical studies. Drugs R D 2016, 16, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Gillen, M.; Millson, D.; Oliver, S.; Brealey, C.; Elsby, R.; Baluom, M.; Lau, D.; Mant, T. Effects of fostamatinib on the pharmacokinetics of digoxin (a P-glycoprotein substrate): Results from in vitro and phase I clinical studies. Clin. Ther. 2015, 37, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Gillen, M.; Ritter, J.; Mathews, D.; Brealey, C.; Surry, D.; Oliver, S.; Holmes, V.; Severin, P.; Elsby, R. Effects of fostamatinib on the pharmacokinetics of oral contraceptive, warfarin, and the statins rosuvastatin and simvastatin: Results from phase I clinical studies. Drugs R D 2016, 16, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.; Gillen, M.; Millson, D.; Oliver, S.; Brealey, C.; Surry, D.; Sweeny, D.; Lau, D.; Leese, P. Effects of fostamatinib on the pharmacokinetics of the CYP2C8 substrate pioglitazone: Results from in vitro and phase 1 clinical studies. Clin. Pharmacol. Drug Dev. 2016, 5, 170–179. [Google Scholar] [CrossRef]
- Martin, P.; Oliver, S.; Gillen, M.; Marbury, T.; Millson, D. Pharmacokinetic properties of fostamatinib in patients with renal or hepatic impairment: Results from 2 phase I clinical studies. Clin. Ther. 2015, 37, 2823–2836. [Google Scholar] [CrossRef]
- Bussel, J.B. Avatrombopag. Br. J. Haematol. 2018, 183, 342–343. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Lin, L.; Yao, H.; Ji, O.; Shen, Q. Therapeutic options for adult patients with previously treated immune thrombocytopenia—A systematic review and network meta-analysis. Hematology 2019, 24, 290–299. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Kuter, D.J. Relative potency of the thrombopoietin receptor agonists eltrombopag, avatrombopag and romiplostim in a patient with chronic immune thrombocytopenia. Br. J. Haematol. 2018, 183, 168. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.; Violi, F. Thrombopoietin receptor agonists and risk of portal vein thrombosis in patients with liver disease and thrombocytopenia: A meta-analysis. Dig. Liver Dis. 2019, 51, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.R.; Koprowski, S.; Hum, J.; McCarty, O.J.T.; DeLoughery, T.G.; Shatzel, J.J. Chronic liver disease, thrombocytopenia and procedural bleeding risk; are novel thrombopoietin mimetics the solution? Platelets 2018, 13, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Ghanima, W.; Geyer, J.T.; Lee, C.S.; Boiocchi, L.; Imahiyerobo, A.A.; Orazi, A.; Bussel, J.B. Bone marrow fibrosis in 66 patients with immune thrombocytopenia treated with thrombopoietin-receptor agonists: A single-center, long-term follow-up. Haematologica 2014, 99, 937–944. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types | Drugs |
|---|---|
| Drug-induced immune thrombocytopenia | Abciximab, Acetaminophen, Amiodarone, Carbamazepine, Ceftriaxone, Daptomycin, Eptifibatide, Ethambutol, Furosemide, Haloperidol, Heparin, Ibuprofen, Irinotecan, Levofloxacin, Mirtazapine, Naproxen, Oxaliplatin, Piperacillin, Phenytoin, Quinidine, Ranitidine, Rifampin, Simvastatin, Sulfonamides including Trimethoprim-sulfamethoxazole, Suramin, Tirofiban, Vancomycin |
| Dose-dependent bone marrow suppression | Daptomycin, Gold compounds, Linezolid, Valproic acid |
| Predicted Properties | Lusutrombopag (3) | Avatrombopag (4) | Fostamatinib (5) |
|---|---|---|---|
| LogP * | 8.08 | 7.09 | 3.24 |
| LogS * | −8.805 | −8.347 | −5.03 |
| pKa * | 3.778 | 4.501 and 8.127 | 1.46 and 2.71 |
| Polar surface area * | 97.22 | 100.84 | 185.13 |
| Refractivity * | 158.17 | 170.04 | 136.52 |
| H-Bond acceptor | 6 | 8 | 13 |
| H-Bond donor | 2 | 2 | 4 |
| Rotatable bonds | 13 | 7 | 10 |
| Number of rings | 3 | 6 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clemons Bankston, P.; Al-Horani, R.A. New Small Molecule Drugs for Thrombocytopenia: Chemical, Pharmacological, and Therapeutic Use Considerations. Int. J. Mol. Sci. 2019, 20, 3013. https://doi.org/10.3390/ijms20123013
Clemons Bankston P, Al-Horani RA. New Small Molecule Drugs for Thrombocytopenia: Chemical, Pharmacological, and Therapeutic Use Considerations. International Journal of Molecular Sciences. 2019; 20(12):3013. https://doi.org/10.3390/ijms20123013
Chicago/Turabian StyleClemons Bankston, Page, and Rami A. Al-Horani. 2019. "New Small Molecule Drugs for Thrombocytopenia: Chemical, Pharmacological, and Therapeutic Use Considerations" International Journal of Molecular Sciences 20, no. 12: 3013. https://doi.org/10.3390/ijms20123013
APA StyleClemons Bankston, P., & Al-Horani, R. A. (2019). New Small Molecule Drugs for Thrombocytopenia: Chemical, Pharmacological, and Therapeutic Use Considerations. International Journal of Molecular Sciences, 20(12), 3013. https://doi.org/10.3390/ijms20123013