Exploring the Relationship of Relative Telomere Length and the Epigenetic Clock in the LipidCardio Cohort

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Relative Leukocyte Telomere Length in LipidCardio
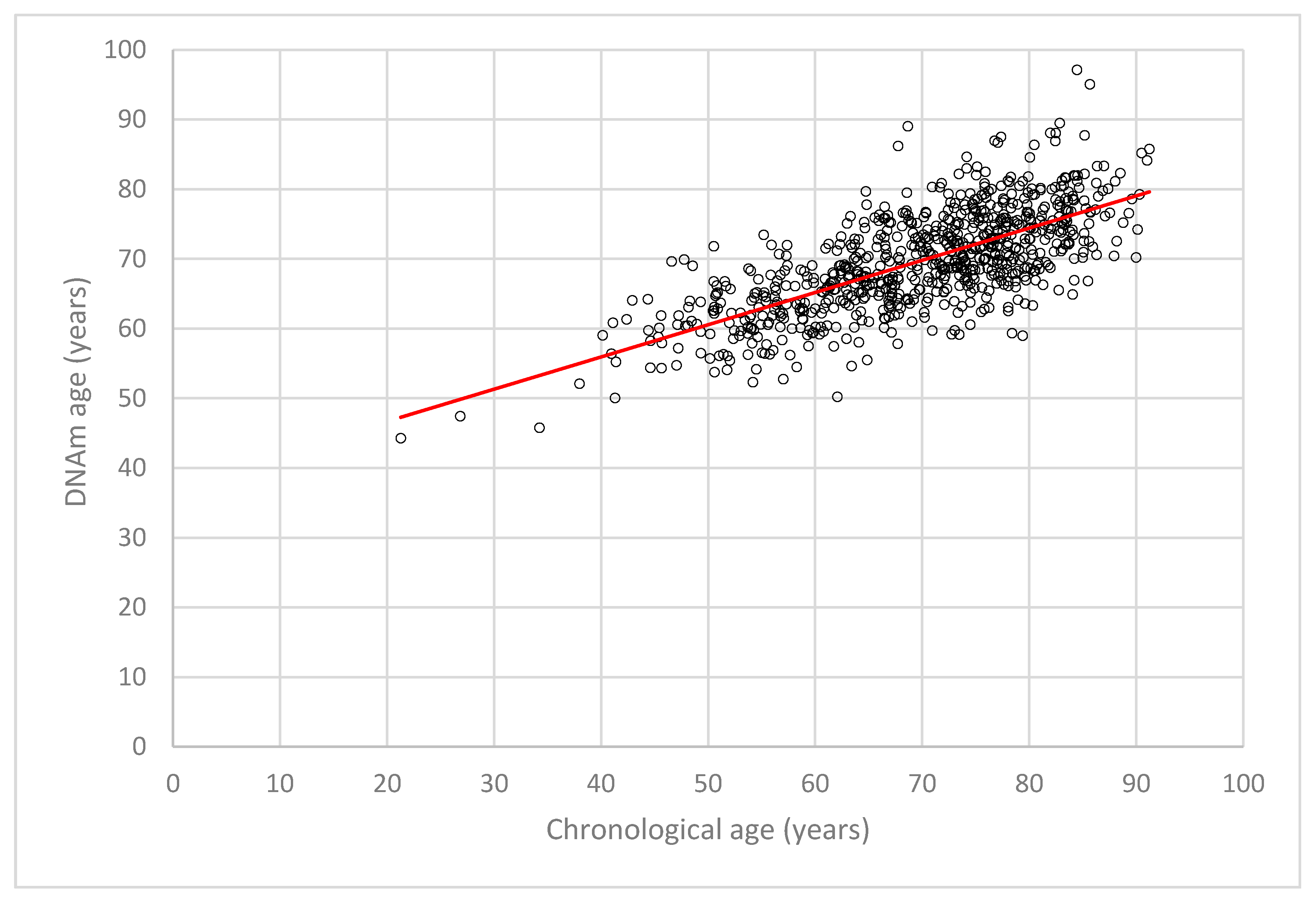
2.2. DNA Methylation (DNAm) Age Estimation and DNAm Age Acceleration in LipidCardio
2.3. Comparison of Different DNA Methylation (DNAm) Age Acceleration Estimates in the Berlin Aging Study II (BASE-II)
2.4. Accuracy of Chronological Age Prediction by the Seven Cytosine-Phosphate-Guanine Sites (CpGs) Epigenetic Clock Across Age Groups
2.5. Relative Leukocyte Telomere Length (rLTL), DNA Methylation (DNAm) Age, and DNAm Age Acceleration in the LipidCardio Cohort
3. Discussion
3.1. Relative Leukocyte Telomere Length and Chronological Age
3.2. The Seven Cytosine-Phosphate-Guanine Sites (CpGs) Epigenetic Clock
3.3. DNAm Age Acceleration
3.4. Relative Leukocyte Telomere Length (rLTL), DNA Methylation (DNAm) Age and DNAm Age Acceleration
4. Materials and Methods
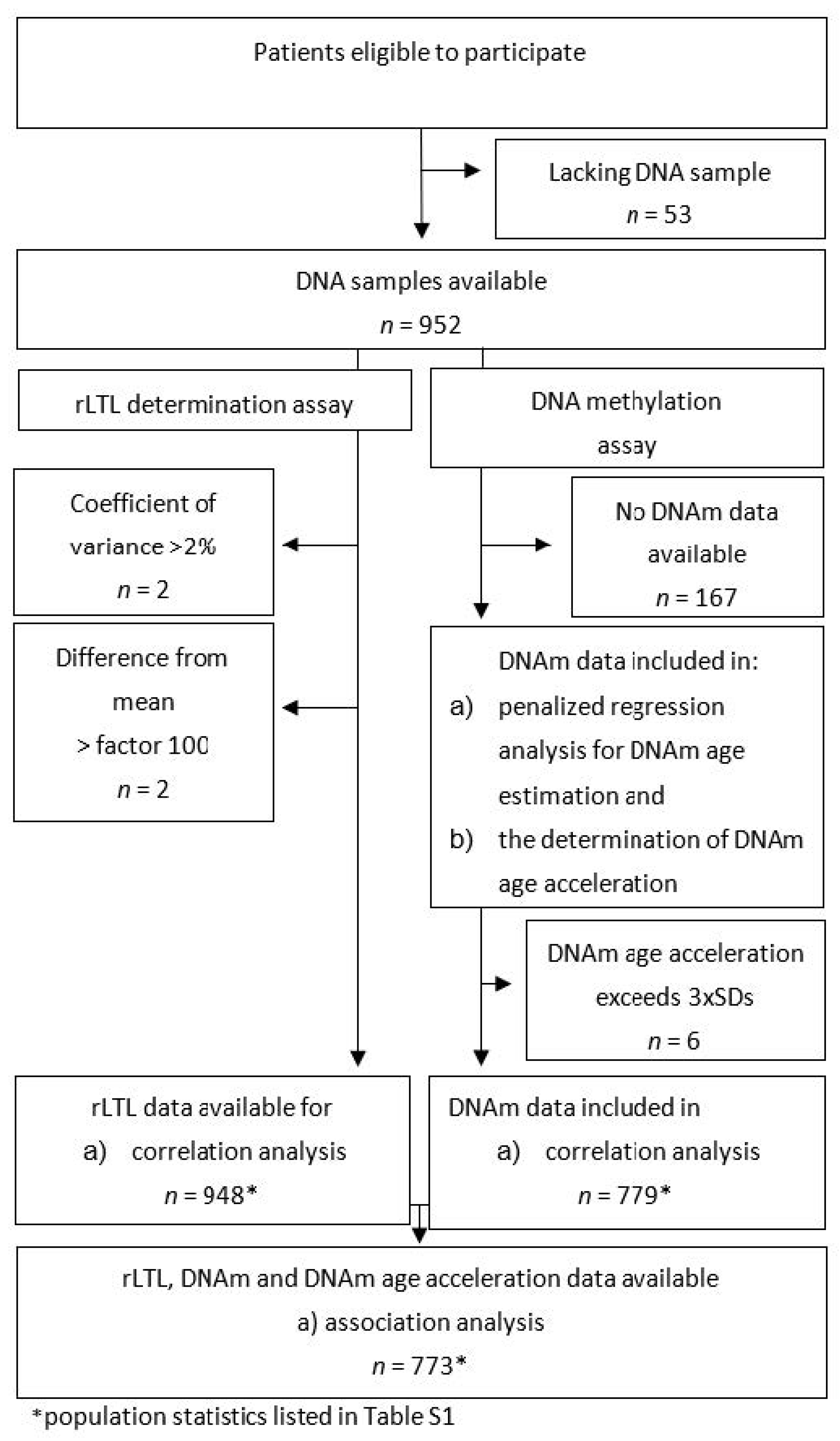
4.1. Missing Data
4.2. Population Characteristics
4.3. DNA Extraction
4.4. Determination of Relative Leukocyte Telomere Length
4.5. DNA Methylation Assay
4.6. Determination of DNA Methylation (DNAm) Age
4.7. Determination of DNA Methylation (DNAm) Age Acceleration
4.8. Exploring the Relationship Between Relative Leukocyte Telomere Length (rLTL) and Epigenetic Clock Variables
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CpG | cytosine-phosphate-guanine |
| CpGs | cytosine-phosphate-guanine sites |
| DNA | Deoxyribonucleic acid |
| DNAm | DNA methylation |
| EEAA | cell-extrinsic epigenetic age acceleration |
| IEAA | cell-intrinsic epigenetic age acceleration |
| PCR | Polymerase chain reaction |
| rLTL | Relative leukocyte telomere length |
| SNuPE | Single nucleotide polymorphism extension |
Appendix A

References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Harley, C.; Futcher, B.; Greider, C. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Tsurimoto, T.; Ishikawa, F. In Vitro Reconstitution of the End Replication Problem. Mol. Cell. Biol. 2001, 21, 5753–5766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Mello, M.; Ross, S.; Briel, M.; Anand, S.; Gerstein, H.; Paré, G. Association Between Shortened Leukocyte Telomere Length and Cardiometabolic Outcomes Systematic Review and Meta-Analysis. Circ. Cardiovasc. Genet. 2015, 8, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C.; Heydon, E.E. Leucocyte telomere length and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2014, 349, g4227. [Google Scholar] [CrossRef]
- Strandberg, T.; Strandberg, A.; Saijonmaa, O.; Tivis, R.; Pitkälä, K.; Fyhrquiat, F. Association between alcohol consumption in healthy midlife and telomere length in older men. The Helsinki Businessmen Study. Eur. J. Epidermiol. 2012, 27, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, T.; Saijonmaa, O.; Tilvis, R.; Strandberg, A.; Miettinen, T.; Pitkälä, K.; Salomaa, V.; Fyhrquist, F. Telomere Length in old age and cholesterol across the life course. J. Am. Geriatr. Soc. 2011, 59, 1979–1981. [Google Scholar] [CrossRef]
- Demissie, S.; Levy, D.; Benjamin, E.J.; Cupples, L.A.; Gardner, J.P.; Herbert, A.; Kimura, M.; Larson, M.G.; Meigs, J.B.; Keaney, J.F.; et al. Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell 2006, 5, 325–530. [Google Scholar] [CrossRef]
- Müezzinler, A.; Zaineddin, A.; Brenner, H. Body mass index and leukocyte telomere length in adults: A systematic review and meta-analysis. Obesity 2013, 15, 192–201. [Google Scholar] [CrossRef]
- Cheng, S.; Yeh, F.; Lin, J.; Matsuguchi, T.; Blackburn, E.; Lee, E.; Howard, B.V.; Zhao, J. Short leukocyte telomere length is associated with obesity in American Indians: The strong heart family study. Aging 2014, 6, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Cherkas, L.; Hunkin, J.; Kato, B.; Richards, B.; Gardner, J.; Surdulescu, G.; Kimura, M.; Lu, X.; Spector, T.D.; Aviv, A. The Association Between Physical Activity in Leisure Time and Leukocyte Telomere Length. Am. Med. Assoc. 2008, 168, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Bralo, L.; Lopez-Golan, Y.; Gonzalez, A. Simplified Assay for Epigenetic Age Estimation in Whole Blood of Adults. Front. Genet. 2016, 7, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jöckel, K.H.; Erbel, R.; Mühleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Vetter, V.; Meyer, A.; Karbasiyan, M.; Steinhagen-Thiessen, E.; Hopfenmüller, W.; Demuth, I. Epigenetic Clock and Relative Telomere Length Represent Largely Different Aspects of Aging in the Berlin Aging Study II (BASE-II). J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.; Harris, S.; Shah, S.; McRae, A.; von Zglinicki, T.; Martin-Ruiz, C.; Wray, N.R.; Visscher, P.M.; Deary, I.J. The epigenetic clock and telomere length are independently associated with chronological age and mortality. Int. J. Epidemiol. 2016, 45, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Belsky, D.; Moffitt, T.; Cohen, A.; Corcoran, D.; Levine, M.; Prinz, J.; Schaefer, J.; Sugden, K.; Williams, B.; Poulton, R.; et al. Eleven Telomere, Epigenetic Clock, and Biomarker-Composite Quantifications of Biological Aging: Do They Measure the Same Thing? Am. J. Epidemiol. 2017, 187, 1220–1230. [Google Scholar] [CrossRef]
- Breitling, L.; Saum, K.-U.; Perna, L.; Schöttker, B.; Holleczek, B.; Brenner, H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin. Epigenet. 2016, 8, 21. [Google Scholar] [CrossRef]
- Weidner, C.I.; Costa, I.G.; Marioni, R.E.; Marcelo, R.P. DNA methylation levels at individual age—Associated CpG sites can be indicative for life expectancy. Aging 2016, 8, 394–401. [Google Scholar]
- Jylhävä, J.; Pedersen, N.; Hägg, S. Biological Age Predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quach, A.; Levine, M.; Tanaka, T.; Lu, A.; Chen, B.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Levine, A.J. HIV-1 Infection Accelerates Age According to the Epigenetic Clock. J. Infect. Dis. 2015, 212, 1563–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schönfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.C.; Spector, T.D.; et al. Obesity accelerates epigenetic aging of human liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, L.; Ingelsson, E.; Sundstr, J.; Siegbahn, A.; Lampa, E. Methylation-based estimated biological age and cardiovascular disease. Eur. J. Clin. Investig. 2018, 48, e12872. [Google Scholar] [CrossRef] [PubMed]
- König, M.; Joshi, S.; Leistner, D.; Landmesser, U.; Sinning, D.; Steinhagen-Thiessen, E.; Demuth, I. Cohort Profile: The LipidCardio Study—Role of Lipoproteins in Cardiovascular Disease. bioRxiv 2019. [Google Scholar] [CrossRef]
- Vidal-Bralo, L.; Lopez-Golan, Y.; Gonzalez, A. Corrigendum: Simplified Assay for Epigenetic Age Estimation in Whole Blood of Adults. Front. Genet. 2017, 8, 51. [Google Scholar] [CrossRef]
- Hastings, W.; Shalev, I.; Belsky, D. Translating Measures of Biological Aging to Test Effectiveness of Geroprotective Interventions: What Can We Learn from Research on Telomeres? Front. Genet. 2017, 8, 164. [Google Scholar] [CrossRef]
- Turner, K.; Vasu, V.; Griffin, D. Telomere Biology and Human Phenotype. Cells 2019, 8, 73. [Google Scholar] [CrossRef]
- Andreassi, M. DNA damage, vascular senescence and atherosclerosis. J. Mol. Med. 2008, 86, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.; Van DerHarst, P. Biological ageing and cardiovascular disease. Heart 2008, 94, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.; Bann, D.; Wiley, L.; Cooper, R.; Hardy, R.; Nitsch, D.; Martin-Ruiz, C.; Shiels, P.; Sayer, A.A.; Barbieri, M.; et al. Gender and telomere length: Systematic review and meta-analysis. Exp. Gerontol. 2015, 51, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Salewsky, B.; Spira, D.; Steinhagen-Thiessen, E.; Norman, K.; Demuth, I. Relative Leukocyte Telomere Length, Hematological Parameters and Anemia—Data from the Berlin Aging Study II (BASE-II). Gerontology 2016, 62, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.; Wright, W.E.; Shay, J.W. Comparison of telomere length measurement methods. Philos. Trans. R. Soc. B 2018, 373, 20160451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinius, L.; Acevedo, N.; Joerinkk, M.; Pershagen, G.; Dahle, S.-E.; Greco, D.; Söderhäll, C.; Scheynius, A.; Kere, J. Differential DNA Methylation in Purified Human Blood Cells: Implications for Cell Lineage and Studies on Disease Susceptibility. PLoS ONE 2012, 7, e41361. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, A.; Irizarry, R. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014, 15, R31. [Google Scholar] [CrossRef]
- Chen, B.; Marioni, R.; Colocino, E.; Peters, M.; Wars-Caviness, C.; Tsai, P.-C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef]
- Chen, B.; Carty, C.; Kimura, M.; Kark, J.; Chen, W.; Li, S.; Zhang, T.; Kooperberg, C.; Levy, D.; Assimes, T.; et al. Leukocyte telomere length, T cell composition and DNA methylation age. Leukoc Telomere Length, T Cell Compos DNA Methylation Age. Aging 2017, 9, 1983–1995. [Google Scholar] [CrossRef]
- Cawthon, R. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002, 30, e47. [Google Scholar] [CrossRef]
- Pfaffl, M. A new mathematical model for relative quantification in real-time RT—PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.; Assadzadeh, A.; Flanagan, J.; Petronis, A. Single nucleotide extension technology for quantitative site-specific evaluation of met C/C in GC-rich regions. Nucleic Acids Res. 2005, 33, e95. [Google Scholar] [CrossRef] [PubMed]
- Rosen, A.; Robertson, K.; Hlady, R.; Muench, C.; Horvath, S.; Kaminsky, Z.; Lohoff, F.W.; Lee, J.; Philibert, R. DNA methylation age is accelerated in alcohol dependence. Transl. Psychiatry 2018, 8, 182. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Variables | Mean ± SD (n) or n and Percentage |
|---|---|
| Number of observations (n) | 773 |
| Female | 244 (31.6%) |
| Chronological age (years) | 69.68 ± 11.01 |
| DNAm age (years) | 69.67 ± 7.27 |
| DNAm age acceleration/ residuals (years) | −0.01 ± 7.83 |
| rLTL | 0.79 ± 0.14 |
| BMI | 27.8 ± 4.8 (704) |
| Diabetes mellitus type II | 208 (26.9%) |
| HDL- cholesterol (mg/dL) | 51.23 ± 16.86 (739) |
| LDL- cholesterol (mg/dL) | 99.28 ± 40.57 (741) |
| Hypertension | 624 (80.7%) |
| Coronary heart disease | 585 (75.8%) (772) |
| Myocardial infarction | 234 (30.4%) |
| Ex-smoker/current smoker | 470 (67.2%) (699) |
| Pack years | 30.15 ± 28.9 (463) |
| Alcohol consumers | 387 (56.0%) (691) |
| Alcohol consumed per week (units) | 5.2 ± 6.2 |
| CpG Site | β | SE | p-Value |
|---|---|---|---|
| Intercept | 101.629 | 2.741 | 5.03 × 10−174 |
| cg19761273 | −77.395 | 10.387 | 2.47 × 10−13 |
| cg17471102 | −27.062 | 4.711 | 1.33 × 10−8 |
| cg02228185 | −20.098 | 2.699 | 2.59 × 10−13 |
| cg09809672 | −18.923 | 4.178 | 7.00 × 10−6 |
| cg10917602 | −7.319 | 2.798 | 9.08 × 10−3 |
| cg16386080 | 43.745 | 6.726 | 1.40 × 10−10 |
| cg24768561 | 8.232 | 4.071 | 4.35 × 10−2 |
| cg25809905 was excluded | |||
| DNAm Age | DNAm Age Acceleration | |||
|---|---|---|---|---|
| (Co-)Variables | β | p-Value | β | p-Value |
| Model 1: rLTL | 4.69 | 0.14 | 1.69 | 0.59 |
| Model 2: rLTL, Chronological age | 3.30 | 0.14 | 3.07 | 0.17 |
| Model 3: rLTL, Chronological age, Sex | 3.00 | 0.18 | 2.74 | 0.22 |
| Model 4: rLTL, Chronological age, Sex, Lifestyle factors (smoking, alcohol consumption) | 3.00 | 0.18 | 2.76 | 0.22 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banszerus, V.L.; Vetter, V.M.; Salewsky, B.; König, M.; Demuth, I. Exploring the Relationship of Relative Telomere Length and the Epigenetic Clock in the LipidCardio Cohort. Int. J. Mol. Sci. 2019, 20, 3032. https://doi.org/10.3390/ijms20123032
Banszerus VL, Vetter VM, Salewsky B, König M, Demuth I. Exploring the Relationship of Relative Telomere Length and the Epigenetic Clock in the LipidCardio Cohort. International Journal of Molecular Sciences. 2019; 20(12):3032. https://doi.org/10.3390/ijms20123032
Chicago/Turabian StyleBanszerus, Verena L., Valentin M. Vetter, Bastian Salewsky, Maximilian König, and Ilja Demuth. 2019. "Exploring the Relationship of Relative Telomere Length and the Epigenetic Clock in the LipidCardio Cohort" International Journal of Molecular Sciences 20, no. 12: 3032. https://doi.org/10.3390/ijms20123032
APA StyleBanszerus, V. L., Vetter, V. M., Salewsky, B., König, M., & Demuth, I. (2019). Exploring the Relationship of Relative Telomere Length and the Epigenetic Clock in the LipidCardio Cohort. International Journal of Molecular Sciences, 20(12), 3032. https://doi.org/10.3390/ijms20123032