Genome-Wide Identification, Expression Analysis, and Subcellular Localization of Carthamus tinctorius bHLH Transcription Factors

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Genome-Wide Identification and Phylogenetic Analysis of Safflower bHLH Genes
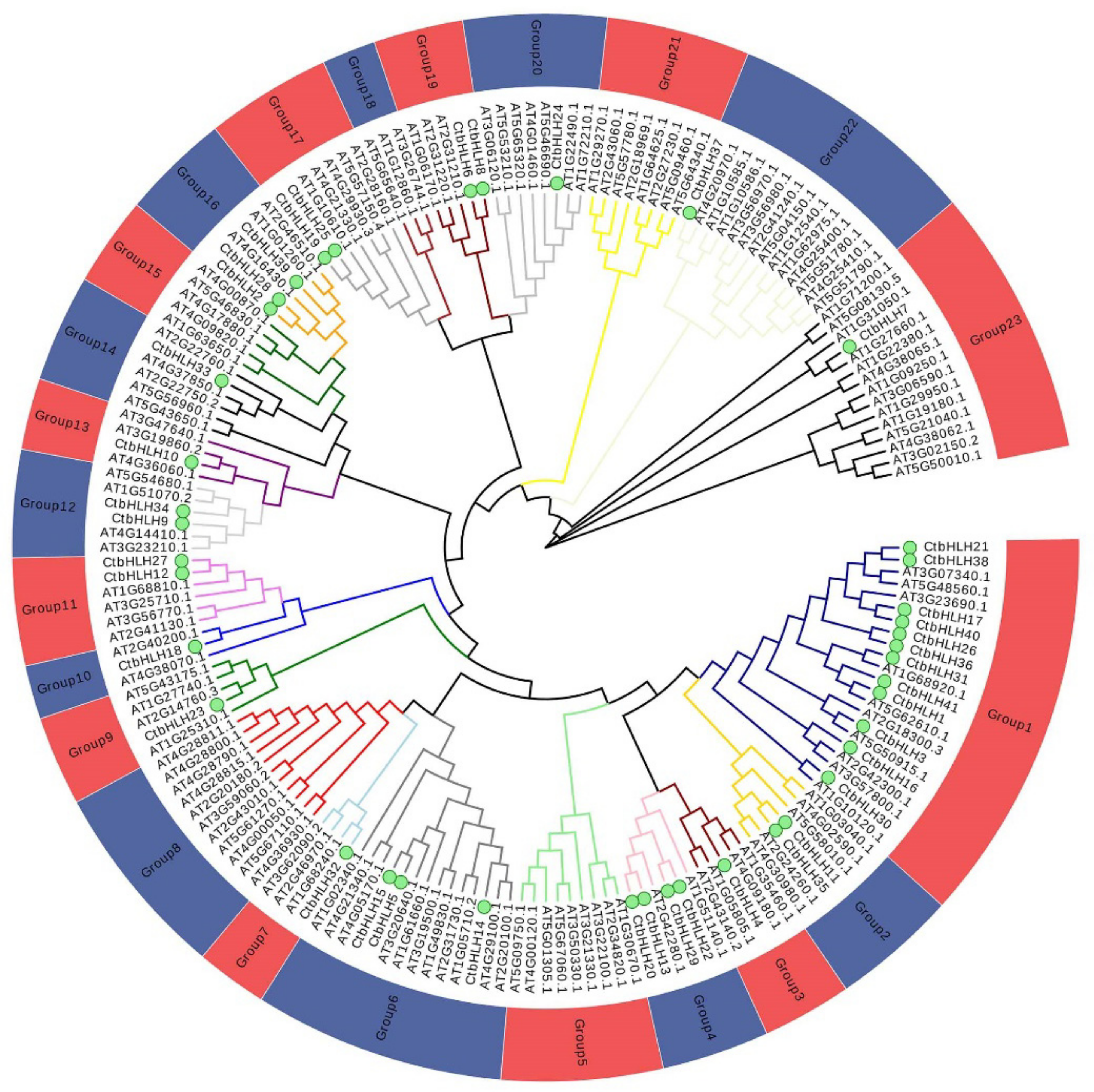
2.2. Phylogenetic Analysis
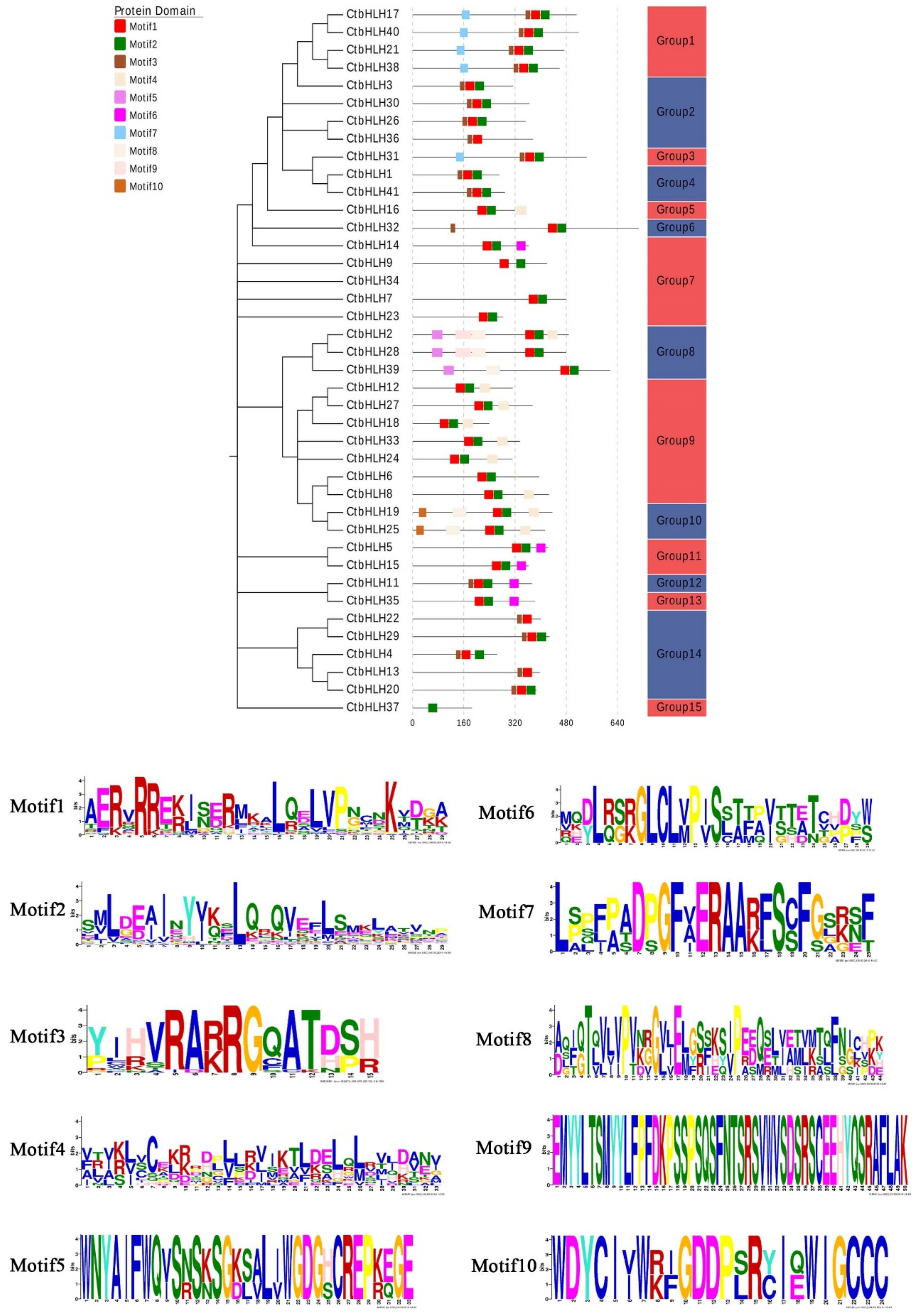
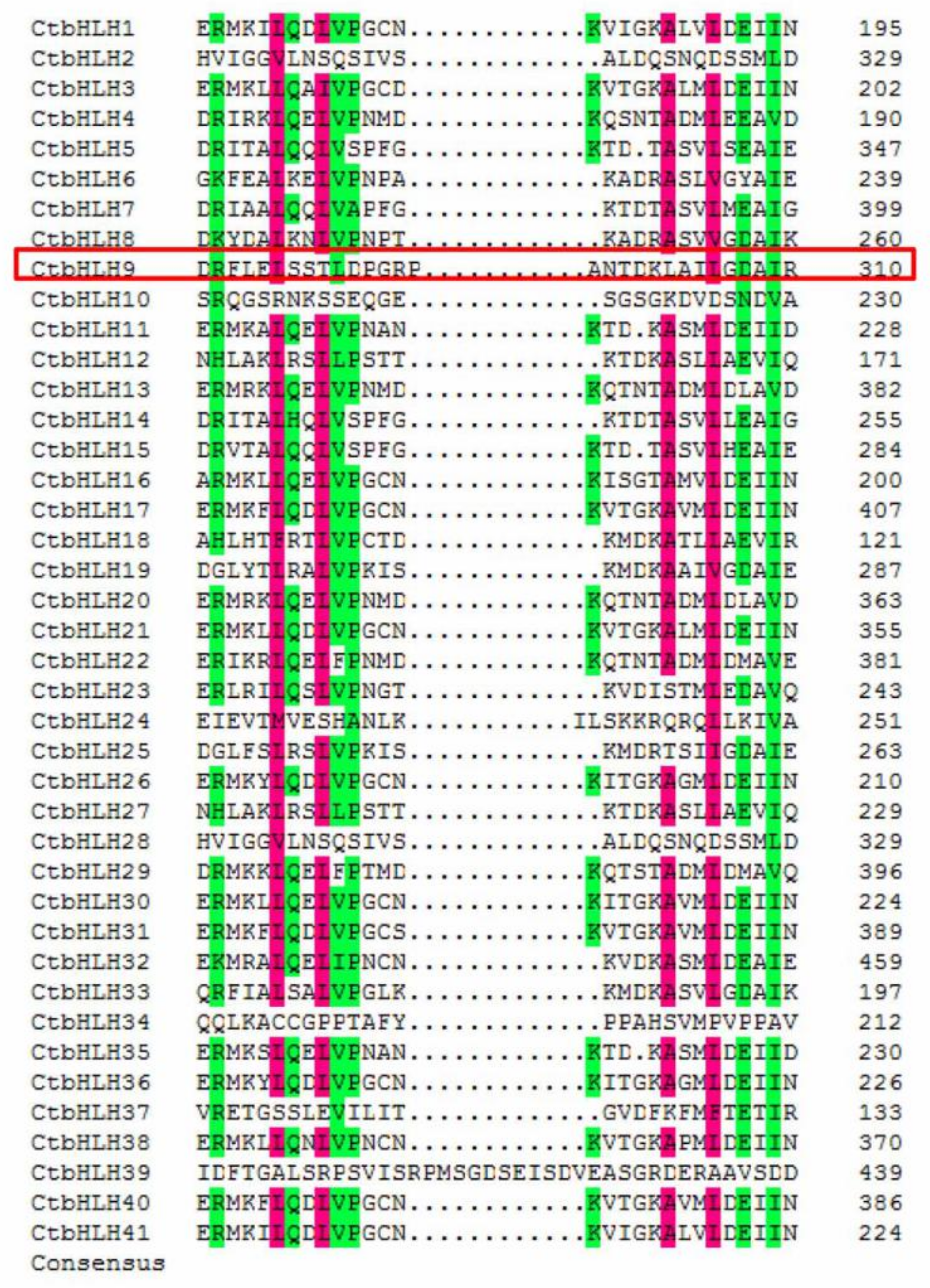
2.3. Motifs and Multiple Sequence Alignment of bHLH Proteins
2.4. Gene Expression Analysis
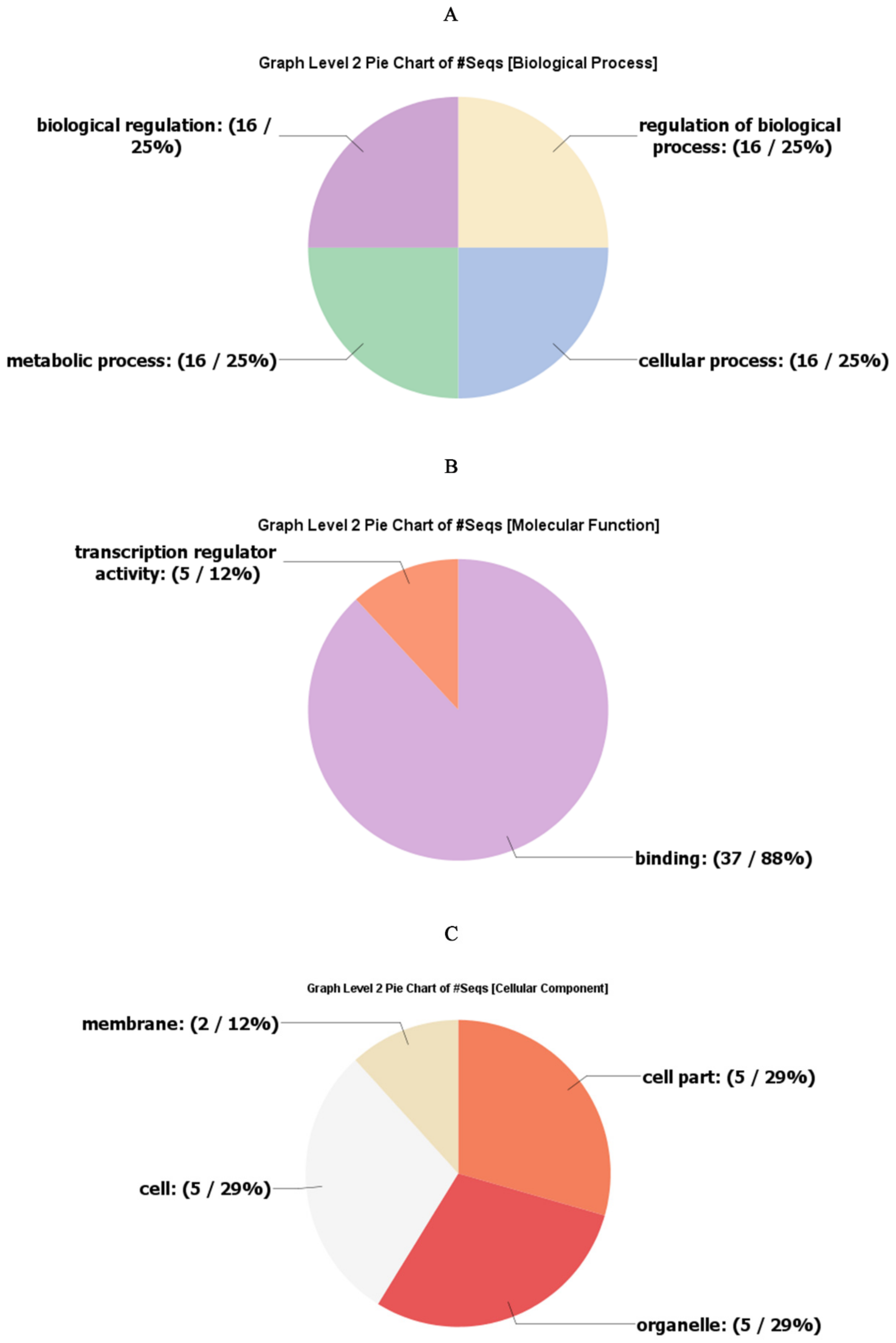
2.5. Gene Ontology Annotations
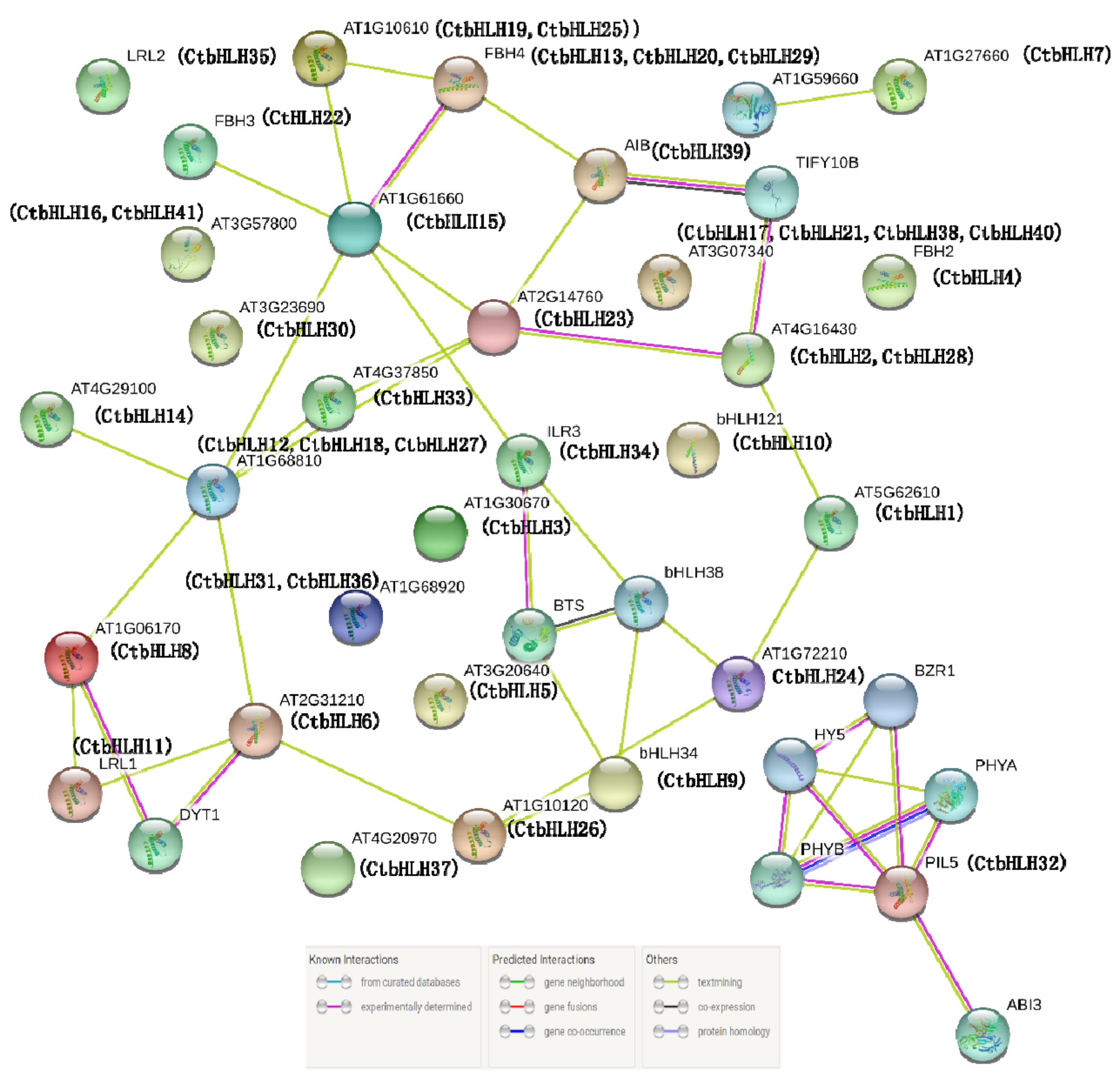
2.6. Protein Clustering Networks
2.7. Topology of Conserved Cis-Elements within the Promoter Regions of CtbHLH Genes
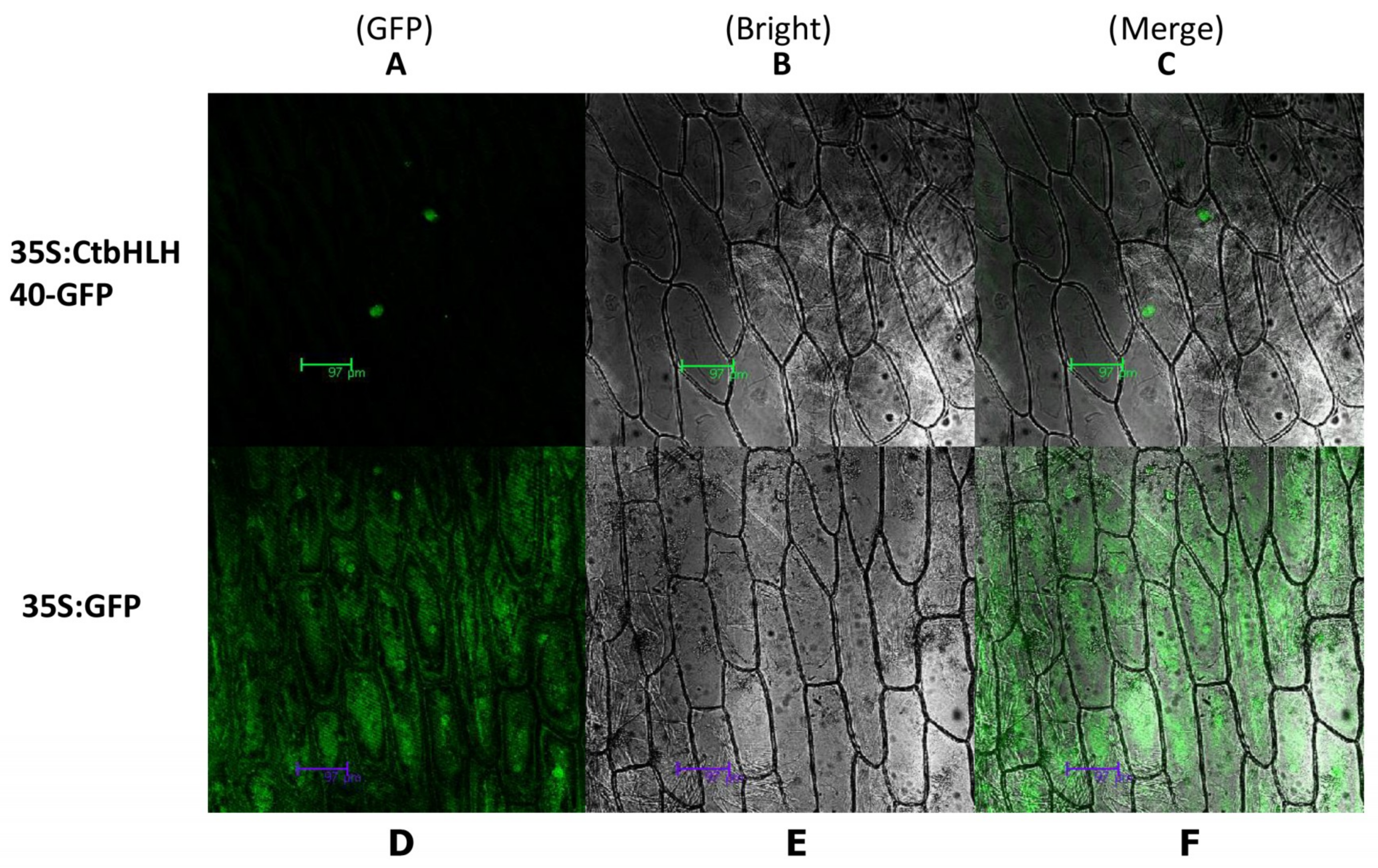
2.8. Subcellular Localization of CmbHLH40
3. Materials and Methods
3.1. Plant Materials
3.2. Identification of Safflower bHLH Superfamily Genes
3.3. Motif Elicitation and Phylogenetic Analysis of bHLH Proteins
3.4. Gene Ontology Annotation
3.5. Expression Analysis of the Carthamus bHLH Protein Superfamily
3.6. Protein Interaction Network Prediction and Functional Annotations Using STRING
3.7. Promoter Sequence Analysis of CtbHLH
3.8. Subcellular Localization of CtbHLH40
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TFs | Transcription factors |
| bHLH | Basic helix–loop–helix |
| CLA | Conjugated linoleic acid |
| GRAVY | Grand average of hydropathicity |
| PI | Isoelectric points |
| Kda | Killi dalton |
| CDS | Coding sequences |
| HMM | Hidden Markov model |
| qRT-PCR | Quantitative real-time PCR |
| MW | Molecular weight |
| TSS | Transcription start site |
| GDB | Genome database |
| CDD | Conserved Domain |
References
- Massari, M.E.; Murre, C. Helix-loop-helix proteins: Regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 2000, 20, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Guan, Y.; Chen, S.; Li, H. Genome-wide analysis of basic helix-loop-helix (bHLH) transcription factors in Brachypodium distachyon. Bmc Genom. 2017, 18, 619. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Ortiz, G.; Huq, E.; Quail, P.H. The Arabidopsis basic/helix-loop-helix transcription factor family. Plant Cell 2003, 15, 1749–1770. [Google Scholar] [CrossRef]
- Li, X.; Duan, X.; Jiang, H.; Sun, Y.; Tang, Y.; Yuan, Z.; Guo, J.; Liang, W.; Chen, L.; Yin, J. Genome-wide analysis of basic/helix-loop-helix transcription factor family in rice and Arabidopsis. Plant Physiol. 2006, 141, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Feller, A.; Machemer, K.; Braun, E.L.; Grotewold, E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011, 66, 94–116. [Google Scholar] [CrossRef] [PubMed]
- Pires, N.; Dolan, L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol. 2009, 27, 862–874. [Google Scholar] [CrossRef]
- Buck, M.J.; Atchley, W.R. Phylogenetic analysis of plant basic helix-loop-helix proteins. J. Mol. Evol. 2003, 56, 742–750. [Google Scholar] [CrossRef]
- Carretero-Paulet, L.; Galstyan, A.; Roig-Villanova, I.; Martínez-García, J.F.; Bilbao-Castro, J.R.; Robertson, D.L. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 2010, 153, 1398–1412. [Google Scholar] [CrossRef]
- Gao, C.; Sun, J.; Wang, C.; Dong, Y.; Xiao, S.; Wang, X.; Jiao, Z. Genome-wide analysis of basic/helix-loop-helix gene family in peanut and assessment of its roles in pod development. Plos ONE 2017, 12, e0181843. [Google Scholar] [CrossRef]
- Yang, S.; LIU, H.-d.; Qiang, Z.; ZHANG, H.-j.; ZHANG, Z.-d.; LI, Y.-d.; WANG, H.-b.; LIU, F.-z. High-throughput sequencing of highbush blueberry transcriptome and analysis of basic helix-loop-helix transcription factors. J. Integr. Agric. 2017, 16, 591–604. [Google Scholar]
- Sun, H.; Fan, H.-J.; Ling, H.-Q. Genome-wide identification and characterization of the bHLH gene family in tomato. Bmc Genom. 2015, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Song, X.-M.; Huang, Z.-N.; Duan, W.-K.; Ren, J.; Liu, T.-K.; Li, Y.; Hou, X.-L. Genome-wide analysis of the bHLH transcription factor family in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol. Genet. Genom. 2014, 289, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Dong, Q.; Li, C.; Liu, C.; Ma, F. Genome wide identification and characterization of apple bHLH transcription factors and expression analysis in response to drought and salt stress. Front. Plant Sci. 2017, 8, 480. [Google Scholar] [CrossRef] [PubMed]
- Ogo, Y.; Nakanishi Itai, R.; Nakanishi, H.; Kobayashi, T.; Takahashi, M.; Mori, S.; Nishizawa, N.K. The rice bHLH protein OsIRO2 is an essential regulator of the genes involved in Fe uptake under Fe-deficient conditions. Plant J. 2007, 51, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Kavas, M.; Baloğlu, M.C.; Atabay, E.S.; Ziplar, U.T.; Daşgan, H.Y.; Ünver, T. Genome-wide characterization and expression analysis of common bean bHLH transcription factors in response to excess salt concentration. Mol. Genet. Genom. 2016, 291, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Luo, H.; Xu, Z.; Zhu, Y.; Ji, A.; Song, J.; Chen, S. Genome-wide characterisation and analysis of bHLH transcription factors related to tanshinone biosynthesis in Salvia miltiorrhiza. Sci. Rep. 2015, 5, 11244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szecsi, J.; Joly, C.; Bordji, K.; Varaud, E.; Cock, J.M.; Dumas, C.; Bendahmane, M. BIGPETALp, a bHLH transcription factor is involved in the control of Arabidopsis petal size. Embo J. 2006, 25, 3912–3920. [Google Scholar] [CrossRef] [Green Version]
- Roh, J.S.; Han, J.Y.; Kim, J.H.; Hwang, J.K. Inhibitory effects of active compounds isolated from safflower (Carthamus tinctorius L.) seeds for melanogenesis. Biol. Pharm. Bull. 2004, 27, 1976–1978. [Google Scholar] [CrossRef]
- Li, X.Z.; Long, R.J.; Yan, C.G.; Lee, H.G.; Kim, Y.J.; Song, M.K. Rumen microbial response in production of CLA and methane to safflower oil in association with fish oil or/and fumarate. Anim. Sci. J. 2011, 82, 441–450. [Google Scholar] [CrossRef]
- Cazzato, E.; Laudadio, V.; Corleto, A.; Tufarelli, V. Effects of harvest date, wilting and inoculation on yield and forage quality of ensiling safflower (Carthamus tinctorius L.) biomass. J. Sci. Food Agric. 2011, 91, 2298–2302. [Google Scholar] [CrossRef]
- Thao, N.T.P.; Cuong, T.D.; Hung, T.M.; Lee, J.H.; Na, M.; Son, J.K.; Jung, H.J.; Fang, Z.; Woo, M.H.; Choi, J.S. Simultaneous determination of bioactive flavonoids in some selected Korean thistles by high-performance liquid chromatography. Arch. Pharmacal Res. 2011, 34, 455. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, J.; Zhou, T.; Guo, B.; Wang, Y.; Hou, J. Simultaneous determination of seven flavonoids in dog plasma by ultra-performance liquid chromatography–tandem mass spectrometry and its application to a bioequivalence study of bioactive components in Herba Epimedii and Er-Xian Decoction. J. Pharm. Biomed. Anal. 2011, 54, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.-Y.; Gao, W.-Y. Hydroxysafflor yellow A protects neuron against hypoxia injury and suppresses inflammatory responses following focal ischemia reperfusion in rats. Arch. Pharmacal Res. 2008, 31, 1010–1015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, J.; Dong, H.; Zhao, X.; Zhou, L.; Li, X.; Liu, J.; Niu, Y. Hydroxysafflor yellow A protects against chronic carbon tetrachloride-induced liver fibrosis. Eur. J. Pharmacol. 2011, 660, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dong, Y.; Yao, N.; Zhang, Y.; Wang, N.; Cui, X.; Li, X.; Wang, Y.; Wang, F.; Yang, J. De Novo Sequencing and Analysis of the Safflower Transcriptome to Discover Putative Genes Associated with Safflor Yellow in Carthamus tinctorius L. Int. J. Mol. Sci. 2015, 16, 25657–25677. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Feng, R.; Ma, R.; Shen, Z.; Cai, Z.; Song, Z.; Peng, B.; Yu, M. Genome-wide analysis of basic helix-loop-helix superfamily members in peach. Plos ONE 2018, 13, e0195974. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-J.; Wang, J.-R. Global identification, structural analysis and expression characterization of bHLH transcription factors in wheat. BMC Plant Biol. 2017, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Li, M.-Y.; Wu, X.-J.; Huang, Y.; Ma, J.; Xiong, A.-S. Genome-wide analysis of basic helix–loop–felix family transcription factors and their role in responses to abiotic stress in carrot. Mol. Breed. 2015, 35, 125. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, P.; Kong, N.; Lu, R.; Pei, Y.; Huang, C.; Ma, H.; Chen, Q. Genome-wide identification and characterization of the potato bHLH transcription factor family. Genes 2018, 9, 54. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. 1994, 2, 28–36. [Google Scholar]
- Heim, M.A.; Jakoby, M.; Werber, M.; Martin, C.; Weisshaar, B.; Bailey, P.C. The basic helix–loop–helix transcription factor family in plants: A genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 2003, 20, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Cui, Y.; Liu, Y.; Fan, H.; Du, J.; Huang, Z.; Yuan, Y.; Wu, H.; Ling, H.-Q. Requirement and functional redundancy of Ib subgroup bHLH proteins for iron deficiency responses and uptake in Arabidopsis thaliana. Mol. Plant 2013, 6, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Zhu, E.; You, C.; Wang, S.; Cui, J.; Niu, B.; Wang, Y.; Qi, J.; Ma, H.; Chang, F. The DYT 1-interacting proteins b HLH 010, b HLH 089 and b HLH 091 are redundantly required for A rabidopsis anther development and transcriptome. Plant J. 2015, 83, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Calvo, P.; Chini, A.; Fernández-Barbero, G.; Chico, J.-M.; Gimenez-Ibanez, S.; Geerinck, J.; Eeckhout, D.; Schweizer, F.; Godoy, M.; Franco-Zorrilla, J.M. The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 2011, 23, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Nesi, N.; Debeaujon, I.; Jond, C.; Pelletier, G.; Caboche, M.; Lepiniec, L. The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. Plant Cell 2000, 12, 1863–1878. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, B.; Deyholos, M.K. Functional characterization of the Arabidopsis bHLH92 transcription factor in abiotic stress. Mol. Genet. Genom. 2009, 282, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Liu, F.; Guo, D.; Fan, L.; Zhu, Z.; Xue, Y.; Gao, Y.; Guo, M. Molecular characterization of flavanone 3-hydroxylase gene and flavonoid accumulation in two chemotyped safflower lines in response to methyl jasmonate stimulation. Bmc Plant Biol. 2016, 16, 132. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Gene ID | Protein Length | PI | MW (Da) | Subcellular Localization | Instability Index | Arabidopsis Homology | GRAVY |
|---|---|---|---|---|---|---|---|---|
| CtbHLH01(4) | CCG019410.2 | 271 | 7.84 | 29436.75 | Nuclear | 59.80 | AT5G62610.1 | −0.732 |
| CtbHLH02(8) | CCG019793.1 | 488 | 6.85 | 54381.40 | Nuclear | 51.53 | AT4G16430.1 | −0.542 |
| CtbHLH03(2) | CCG020831.1 | 314 | 7.78 | 35586.93 | Nuclear | 46.88 | AT1G30670.1 | −0.770 |
| CtbHLH04(14) | CCG021763.1 | 264 | 6.66 | 29051.21 | Nuclear | 43.29 | AT4G09180.1 | −0.272 |
| CtbHLH05(11) | CCG022142.1 | 424 | 5.63 | 46461.98 | Nuclear | 61.08 | AT3G20640.1 | −0.770 |
| CtbHLH06(9) | CCG023511.1 | 396 | 5.54 | 45156.48 | Nuclear | 43.12 | AT2G31210.1 | −0.620 |
| CtbHLH07(7) | CCG028074.4 | 480 | 6.73 | 53210.85 | Nuclear | 48.98 | AT1G27660.1 | −0.476 |
| CtbHLH08(9) | CCG028576.1 | 426 | 5.62 | 47924.37 | Nuclear | 37.73 | AT1G06170.1 | −0.654 |
| CtbHLH09(7) | CCG029033.2 | 420 | 9.34 | 48353.34 | Nuclear | 51.47 | AT3G23210.1 | −0.712 |
| CtbHLH10 | CCG029306.2 | 294 | 5.93 | 32779.18 | Nuclear | 67.22 | AT3G19860.2 | −1.017 |
| CtbHLH11(12) | CCG031433.1 | 373 | 5.88 | 38966.22 | Nuclear | 56.54 | AT2G24260.1 | −0.424 |
| CtbHLH12(9) | CCG031433.1 | 313 | 6.64 | 34357.55 | Nuclear | 57.98 | AT1G68810.1 | −0.587 |
| CtbHLH13(14) | CCG003415.1 | 398 | 5.61 | 44597.03 | Nuclear | 61.12 | AT2G42280.1 | −0.826 |
| CtbHLH14(7) | CCG003696.1 | 362 | 6.53 | 39563.23 | Nuclear | 67.71 | AT1G51140.1 | −0.690 |
| CtbHLH15(11) | CCG004828.1 | 363 | 6.33 | 39459.94 | Nuclear | 57.93 | AT1G61660.1 | −0.602 |
| CtbHLH16(5) | CCG006936.1 | 320 | 5.21 | 35518.73 | Nuclear | 60.39 | AT3G57800.1 | −0.585 |
| CtbHLH17(1) | CCG009097.1 | 513 | 5.61 | 56099.86 | Nuclear | 42.63 | AT3G07340.1 | −0.677 |
| CtbHLH18(9) | CCG010966.1 | 249 | 9.15 | 27766.58 | Nuclear | 31.23 | AT1G68810.1 | −0.453 |
| CtbHLH19(10) | CCG015095.1 | 437 | 7.81 | 49777.63 | Nuclear | 34.05 | AT1G10610.1 | −0.390 |
| CtbHLH20(14) | CCG016487.1 | 388 | 5.95 | 43102.83 | Nuclear | 58.48 | AT2G42280.1 | −0.726 |
| CtbHLH21(1) | CCG030113.1 | 474 | 6.34 | 51930.85 | Nuclear | 51.00 | AT3G07340.1 | −0.720 |
| CtbHLH22(14) | CCG000235.1 | 401 | 6.07 | 44439.58 | Nuclear | 69.25 | AT1G51140.1 | −0.913 |
| CtbHLH23(7) | CCG000365.1 | 283 | 5.59 | 31696.50 | Nuclear | 52.80 | AT2G14760.3 | −0.654 |
| CtbHLH24(9) | CCG001914.1 | 312 | 7.05 | 34294.77 | Nuclear | 64.73 | AT1G72210.1 | −0.500 |
| CtbHLH25(10) | CCG002853.1 | 414 | 7.96 | 46510.11 | Nuclear | 41.81 | AT1G10610.1 | −0.434 |
| CtbHLH26(2) | CCG003971.1 | 353 | 6.47 | 39567.82 | Nuclear | 40.90 | AT1G10120.1 | −0.587 |
| CtbHLH27(9) | CCG006284.1 | 375 | 6.37 | 42123.27 | Nuclear | 69.08 | AT1G68810.1 | −0.696 |
| CtbHLH28(8) | CCG006712.1 | 488 | 6.85 | 54381.40 | Nuclear | 52.40 | AT4G16430.1 | −0.548 |
| CtbHLH29(14) | CCG008781.1 | 428 | 6.63 | 48008.56 | Nuclear | 61.50 | AT2G42280.1 | −0.997 |
| CtbHLH30(2) | CCG009709.1 | 364 | 8.13 | 40547.38 | Nuclear | 43.90 | AT3G23690.1 | −0.736 |
| CtbHLH31(3) | CCG010368.1 | 544 | 5.42 | 58619.71 | Nuclear | 57.98 | AT1G68920.1 | −0.712 |
| CtbHLH32(6) | CCG011082.1 | 706 | 5.95 | 77127.87 | Nuclear | 58.02 | AT2G20180.2 | −0.576 |
| CtbHLH33(9) | CCG011764.1 | 336 | 7.72 | 37338.69 | Nuclear | 47.03 | AT4G37850.1 | −0.424 |
| CtbHLH34(7) | CCG013712.2 | 250 | 5.64 | 27865.55 | Nuclear | 59.41 | AT5G54680.1 | −0.614 |
| CtbHLH35(13) | CCG014470.1 | 382 | 5.35 | 39191.40 | Nuclear | 53.49 | AT4G30980.1 | −0.375 |
| CtbHLH36(2) | CCG017266.1 | 376 | 5.46 | 42211.90 | Nuclear | 49.28 | AT1G68920.1 | −0.779 |
| CtbHLH37(15) | CCG018780.1 | 186 | 7.03 | 21187.15 | Nuclear | 44.33 | AT4G20970.1 | −0.465 |
| CtbHLH38(1) | CCG018923.1 | 460 | 6.68 | 50715.86 | Nuclear | 56.68 | AT3G07340.1 | −0.676 |
| CtbHLH39(8) | CCG018976.1 | 617 | 9.04 | 69705.26 | Nuclear | 53.97 | AT2G46510.1 | −0.497 |
| CtbHLH40(1) | CCG019036.1 | 519 | 6.31 | 57019.01 | Nuclear | 44.67 | AT3G07340.1 | −0.682 |
| CtbHLH41(4) | CCG019190.1 | 298 | 5.67 | 32673.94 | Nuclear | 64.80 | AT3G57800.1 | −0.933 |
| CtbHLH01(4) | CCG019410.2 | 271 | 7.84 | 29436.75 | Nuclear | 59.80 | AT5G62610.1 | −0.732 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.; Ahmad, N.; Tian, Y.; Liu, J.; Wang, L.; Wang, G.; Liu, X.; Dong, Y.; Wang, F.; Liu, W.; et al. Genome-Wide Identification, Expression Analysis, and Subcellular Localization of Carthamus tinctorius bHLH Transcription Factors. Int. J. Mol. Sci. 2019, 20, 3044. https://doi.org/10.3390/ijms20123044
Hong Y, Ahmad N, Tian Y, Liu J, Wang L, Wang G, Liu X, Dong Y, Wang F, Liu W, et al. Genome-Wide Identification, Expression Analysis, and Subcellular Localization of Carthamus tinctorius bHLH Transcription Factors. International Journal of Molecular Sciences. 2019; 20(12):3044. https://doi.org/10.3390/ijms20123044
Chicago/Turabian StyleHong, Yingqi, Naveed Ahmad, Yuanyuan Tian, Jianyu Liu, Liyan Wang, Gang Wang, Xiuming Liu, Yuanyuan Dong, Fawei Wang, Weican Liu, and et al. 2019. "Genome-Wide Identification, Expression Analysis, and Subcellular Localization of Carthamus tinctorius bHLH Transcription Factors" International Journal of Molecular Sciences 20, no. 12: 3044. https://doi.org/10.3390/ijms20123044