1. Introduction
Membrane proteins (MPs) are an important type of protein along with soluble globular proteins, fibrous proteins, and disordered proteins [
1]. MPs are involved in various crucial biological functions [
2], such as transportation through membranes, immune system molecule recognition, hormone reception, etc., and this is why they have a strong potential to be the target of new drugs in the future, since over half of modern therapeutic drugs target MPs [
3]. Therefore, exploring the functions of membrane proteins, especially their binding capability, remains to be of profound importance on various fronts, not least of which includes drug discovery [
4]. Currently, commercial drug target discovery mainly relies on traditional approaches such as high throughput screening or functional assays, etc. However, more efficient and economical approaches are required to identify the MP-ligand binding sites (MPLs) in detail for modern medicine. Obviously, intelligent computation is a promising approach for this purpose.
During the past decades, many considerable efforts have been applied towards predicting soluble protein-ligand interactions, and these are classified into three major categories: structure-based, sequence-based, and hybrid methods that use both sequence and structure characteristics. Structure-based methods follow the assumption that proteins with similar functions always have similar global or local structures. As an example, identifying ligand binding sites by using structural modeling and information was widely researched [
5,
6,
7,
8]. Since there are fewer protein structures available for more extensive requirements, sequence-based methods have been used to directly predict the residues that probably interact with a particular ligand [
9,
10,
11,
12,
13]. However, the performance of these methods depends highly on whether the sequence-derived features can describe the differences between binding and non-binding residues. Considerable attention has since been paid to the hybrid method that combines structural and sequential information [
14,
15,
16,
17]. Previous studies demonstrated that hybrid methods are often superior to others because they inherit the advantages from both structure- and sequence-based methods.
Suresh et al. [
18] published the first sequence-based MPL predictor Tm-lig for membrane proteins. However, aside from this study, few peer-reviewed works have been reported, while plentiful attention had been paid to their soluble partners. It is obvious that the challenges remain in the membrane protein MPL prediction, but opportunities also currently exist.
The structure-based approach was not applied to membrane proteins directly in this paper since the 3D structures required for comparative homology predictions are not abundant for membrane proteins compared to soluble proteins, which is caused by the native environment [
19,
20,
21] where the hydrophobic thickness of the lipid bilayer they are inserted into exists [
22]. Consequently, sequence-based features are much more accessible than structure-based features for membrane proteins in many realistic scenarios. Thus, the sequence-based approach is preferred here rather than the structure-based prediction.
As in many typical computational biological modes of research, the imbalanced learning problem exists within MP-ligand binding prediction, where the number of majority samples (non-binding residues) is significantly larger than that of minority samples (binding residues). According to statistics, the number of non-binding residues is about 150–200 times more than of that of binding residues. Numerous studies have shown that directly using the traditional classifier algorithm to imbalanced problems often tend to bias toward the larger classes [
23,
24,
25]. Therefore, the data imbalance phenomenon is an inevitable problem to be solved.
Most of the soluble protein-ligand binding site prediction methods, as well as the only-MPLs prediction method, focus on predicting universal ligand binging sites without considering the differences among various ligands. In fact, significant distinctions exist among the different types of ligands based on their size, structure, function, or other characteristics, and different types of ligands tend to attach with particular residues referenced in the surrounding environment. Many ligand-specific binding site predictors have been developed recently [
26,
27,
28], and these are often superior to universal-purpose binding site predictors. Considering this, in addition to the universal ligand-binding residue prediction model, we further build ligand-specific models to predict drug-like compound-binding, metal-binding, and biomacromolecule-binding residues, respectively.
Finally, we analyze the characteristics of MPLs and the contribution of different types of features in detail. We find that the position-specific scoring matrix (PSSM) features are the most effective. At the same time, combining four types of features could more effective which achieve the highest MCC value. From a biological perspective, the PSSM, topology, evolutionary profiles (PSSM), topology structure (TOPO), physicochemical properties (PCP), and primary sequence segment descriptors (SeqSeg) present the evolutionary information, the fundamental structure, the microenvironment, and the original sequence composition of the ligand binding sites, respectively.
In this study, we developed a membrane protein-ligand binding site predictor MPLs-Pred. The limitations of the structure-based methods motivates us to extract discriminative features of the MPLs from sequence information alone, such as evolutionary profiles, topology structure, physicochemical properties, and primary sequence segment descriptors. To tackle the serious impact of the data imbalance phenomenon, a random under-sampling scheme was applied before using the random forest classifier to predict MPLs. The universal MPLs-Pred derived a 0.63 MCC (Matthews correlation coefficient) value on the independent validation test. Considering the distinction among the different types of ligand-binding residues, we divided the ligand-binding residues into three categories, including drug-like compound-binding, metal-binding, and biomacromolecule-binding residues. We built ligand-specific models to further improve the prediction performance and achieved consideracn the independent validation tests, respectively. MPLs-Pred is freely accessible at
http://icdtools.nenu.edu.cn/.
2. Results and Discussion
2.1. Characteristics of Ligand-Binding Residues
In this work, the ligand-binding residue is coded by the features of evolutionary profiles, topology, physicochemical properties, and primary sequence segment descriptors. Before employing these characteristics as the feature spaces of the predictor, we demonstrated the effectiveness of each by statistical and experimental methods.
Figure 1 shows the relative composition of ligand-binding residues based on the universal training dataset and ligand-specific binding residues on the corresponding dataset. The relative composition might reflect the enrichment and distribution of binding residues. It is observed that polar hydrophilic residues such as cysteine (C), histidine (H), and aspartic acid (D) are more likely to be binding sites for all kinds of ligands, especially for metal-binding residues. Biomacromolecules prefer to interact with alkaline residues, such as histidine (H), arginine (R), and lysine (K), as well as the polar hydrophilic residues. This phenomenon also exists in drug-binding residues but is not as significant as others. The enrichment of metal- and biomacromolecule-binding residues is more obvious than that of drug-binding residues. It is probably because the drugs are manmade chemical compounds and not natural ones. The result of long-term evolution makes the proteins form a special region for recognizing specific natural ligands to interact with and perform specific functions. The phenomenon of the low specificity of drug-binding residues leads to the interaction of chemical drugs and membrane proteins that are not in a perfect one-to-one correspondence; this is why drugs always have side effects. It is obvious that different types of ligand-binding residues show a different preference for residue enrichment. Thus, the introduction of the ligand-specific predict strategy is explicable.
Furthermore, the residue preferences of the neighboring environment of target residues were investigated. Two-sample logos maps of universal ligand-binding residues against corresponding non-binding residues are shown in
Figure 2. According to the illustration, the enrichment phenomenon of neighboring residues of the target is not remarkable, which is probably a reflection of the contribution limits of sequence neighbor residues during the interacting process. Thus, over-introducing the information of neighbor residues may cause noise. This phenomenon is different from the case of soluble proteins, and the detailed reason remains to be further explored.
We further investigated the topology distribution of binding residues on the training dataset. According to the predicted result of HMMTOP, among the 10,143 binding residues, 7978 residues (about 79%) were located on the outer side of the membrane, 1788 residues (about 18%) were located on the inner side of the membrane, and only 377 residues (less than 4%) binding residues were located on the transmembrane region. The reason for this phenomenon is a special function of membrane proteins: transmitted ligand, signal, and energy inside and outside of the cell. The residues located between phospholipids are always stable in order to keep the channel structure of membrane proteins.
2.2. The Contribution of Features
As described in
Section 3.2, “Selected Features”, we employed four kinds of features to construct the feature spaces of the predictor. To evaluate these features, we calculated the Pearson correlation coefficient between features and labeled the universal and ligand-specific training datasets. As shown by the heat map in
Figure 3, the features of PSSM reveal the highest negative correlation with the label. The features of PCP also show significant correlations on all datasets. The features of TOPO and SeqSeg show a lower correlation with labels, which might be because the feature vector was too sparse. In a point-by-point comparison among the universal and ligand-specific datasets, the linear correlation between features and labels is relatively lower on drug-like compound data and significantly higher on metal data. This phenomenon is consistent with the experimental results.
We further analyzed the contribution of different kinds of features to the predictor. As shown in
Table 1. The features of PSSM, which presents the evolutionary information of protein sequences, are the most effective. The sequence conservation is obvious in the binding regions, but not all the conserved sequenced segments contain binding sites. Thus, this feature would be a necessary condition for the prediction (seen from high ACC values) but is not a sufficient condition (seen from low Sen values). We found that the topology feature did not obviously improve the prediction performance for MCC, but it did highly increase the Sen. It means that the topology feature helps the predictor to discover the binding sites in the transmembrane segments, even though the predicted topology is not accurate enough. At the same time, four features can be more sufficient and balanced to exhibit the existence of binding sites with similarly high ACC and the highest Sen, which would lead to the highest MCC. From a biological perspective, topology can present the fundamental aspect of the structure of the transmembrane protein, PCP can present the microenvironment of binding regions, and SeqSeg can present the original sequence composition surrounding the binding sites.
2.3. The Random Under-Sampling Scheme Influences Predictor Performance
Membrane protein-ligand binding site prediction is a typical imbalance problem. As illustrated in
Table 1, the negative samples are about 140–200 times more than the positive ones and will cause considerable noise. In this study, we used the random under-sampling scheme to reduce the negative influence of imbalanced data. Due to the limited number of positive samples, we kept all of them and randomly selected some negative samples to build a sub-training dataset. The ratio of negative and positive samples in the sub-training dataset is the most important parameter that seriously affects the performance of the predictor.
Figure 4 shows the tendency of the MCC value as the ratio changes on (a) the training dataset and (b) the independent testing dataset. We can see in
Figure 4a that the predictor achieves the best MCC value when the ratio is at 1, decreases rapidly as the ratio increases, and tends to stabilize when the ratio is >30. We put forward an inference about this situation: When the ratio is <30, selected negative samples are too small to describe the distribution of the original negative samples, and this may cause serious information lost; when the ratio is ≥30, the MCC value tends to stabilize but decrease smoothly due to the increasing noise of redundant samples. The tendency of the MCC value from independent validation as shown in
Figure 4b further verifies our inference.
2.4. Comparison with other Machine Learning Methods Over Cross-Validation
In this section, we compare the random forest classifier with other classification methods on the training dataset. As illustrated in
Table 2, it is obvious that the ensemble classifier, including AdaBoost and RF, performs better than others. This might be because of the serious imbalance between positive and negative samples, and the ensemble method can reduce the negative influence of imbalanced data. The random forest classifier achieves the best MCC value.
2.5. Performance of MPLs-Pred
The performance of MPLs-Pred on the training datasets over 10-fold cross-validation test are listed in
Table 3. By observing the illustration in
Table 3 and compared with universal models, the ligand specific predictor can significantly improve the predictive effect for metal- and biomacromolecule-binding residues, especially for metal-binding residues. However, the prediction accuracy of drug-like compound-binding residues is much worse than the universal model. We have more interest in why drug-binding residue prediction performs worse than others. We speculate that drugs, which are man-made chemical compounds, are very different from natural ligands. According to the statistics from the previous section, the differences between drug-binding residues and non-drug-binding residues are not as significant as that of natural ligand’s. This is because the process of interaction between the membrane protein and the ligand is interventional: the membrane protein will not evolve a special region to recognize a particular drug and further its binding with it. Thus, features derived from sequences cannot commendably describe the characteristics of drug-binding residues.
In order to prove the robustness of the predictor, we further compared the universal model and ligand-specific models on the independent testing dataset. The details are illustrated in
Table 4. We found that all the ligand-specific predictors performed better than the universal one. The metal-ligand binding sites predictor achieved the best performance because of the highly significant characteristics of metal-binding residues.
2.6. Case Studies
To further demonstrate the effectiveness of the MPLs predictor on the universal model and ligand-specific models, we took a metal-binding (UniProt ID: P00959, PDB ID: 1pfu) protein and a drug-binding protein (UniProt ID: Q43133, PDB ID: 2j1p) in the testing dataset for case studies.
P00959 is a cytoplasm membrane protein in
Escherichia Coli. It is the target protein of ATP, tRNA, and zinc ions, and also participates in aminoacyl-tRNA ligase activity. Four zinc binding sites are annotated in UniProt. The protein structure and zinc ion-binding residues are visualized in
Figure 5a. It is obvious that the four amino acids not contiguous in the sequence are spatially clustered, forming a functional domain. Furthermore, the prediction results generated by MPLs-Pred with the universal model and metal-specific model are also illustrated. The result shows that the metal-specific model outperforms the universal model. MPLs-Pred with the metal-specific model correctly predicted all four binding residues, and the universal model correctly identified three out of the four binding residues.
Q43133 is a chromoplast membrane protein of
Sinapis Alba. It is the target protein of magnesium, isopentenyl diphosphate, and dimethylallyl diphosphate. Six dimethylallyl diphosphate binding sites are annotated in UniProt. The details of the protein structure and drug-binding model are visualized in
Figure 5b. Similar to the previous example, the six binding residues also form a functional domain. MPLs-Pred also achieved considerable accuracy on this protein. The universal model correctly predicted three out of the six binding residues, and the drug-specific model correctly predicted five out of the six binding residues.
2.7. Comparison with Existing Methods on the Independent Testing Dataset
In this study, we proposed a novel method to predict ligand-binding residues in membrane proteins, named MPLs-Pred. In order to verify the generalization capability of MPLs-Pred, we compared it with existing predictors on the independent dataset.
There is only one previous study on the field of membrane protein-ligand binding residue prediction created by M. Xavier Suresh and his team in 2015 [
18]. This predictor is named Tm-lig and is freely accessed from
http://tmbeta-genome.cbrc.jp/tm-lig/tm-lig.html. Tm-lig encodes the residues by PSI-BLAST-generated PSSM profiles and employes the Naïve Bayes classifier to predict the candidate ligand-binding residues in membrane proteins. We compared MPLs-Pred predictor with the Tm-lig predictor on the independent dataset. The details are illustrated in
Table 5.
The testing result proves that MPLs-Pred has better performance than Tm-lig on all the evaluation indexes, especially on the MCC value, which reflects the overall performance of the predictor.
2.8. Homo’s Membrane Protein-Ligand Interactions
We have much interest in Homo’s MP-ligand interactions. Here, we built an exclusive predictor to identify ligand-binding residues in Homo’s membrane proteins and achieved considerable performance with 10-fold cross-validation. The ACC, Spe, Sen, and MCC values were 0.992, 0.993, 0.705, and 0.486, respectively.
In this section, we analyze the gene ontology and pathway of Homo’s drug-binding membrane proteins to help understand their functions.
Gene ontology (GO) is an important initiative to unify the representation of gene and gene product attributes across all species. The enrichment analysis is to test whether a GO term is statistically enriched for any given data.
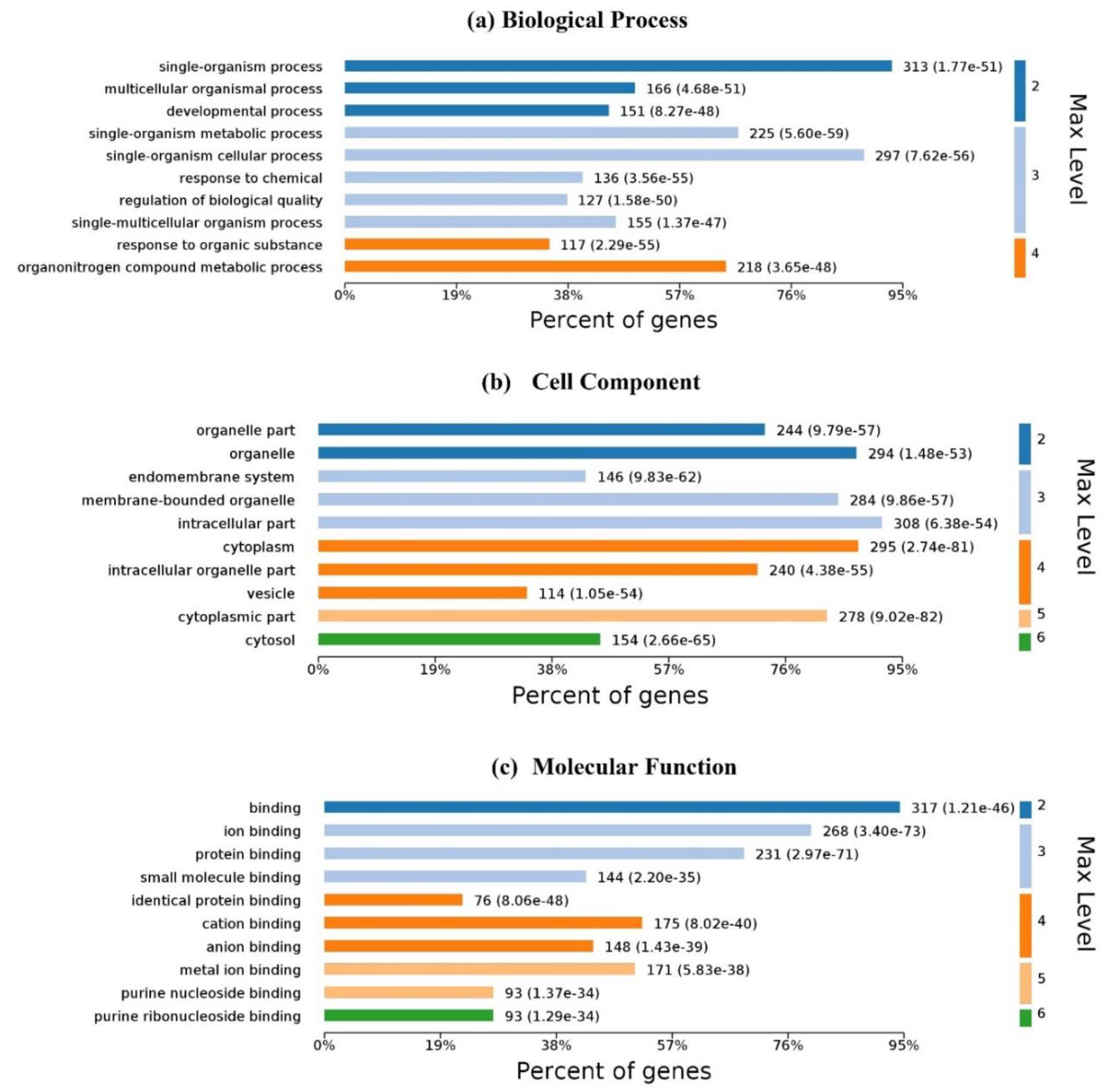
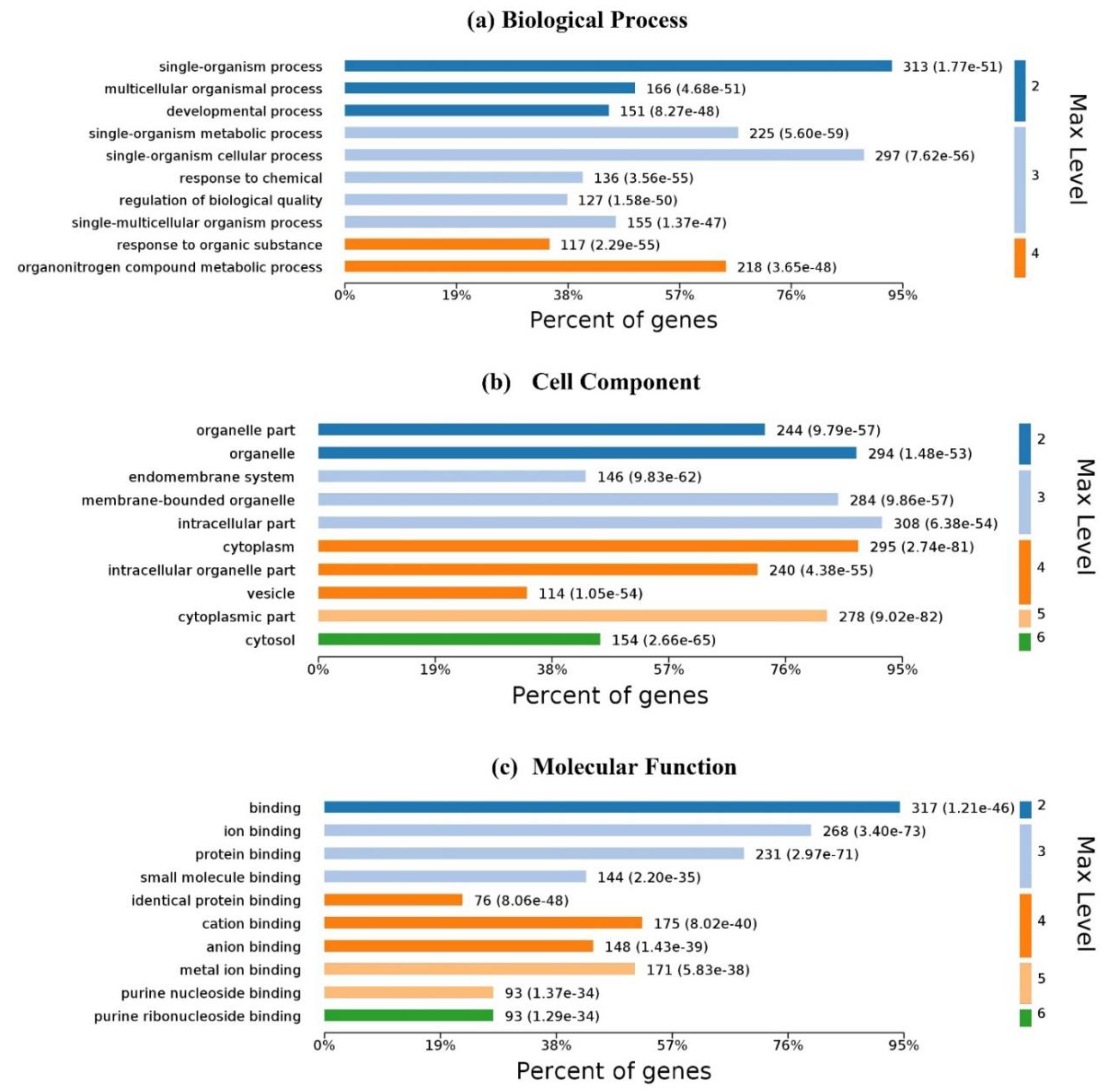
Figure 6 illustrates the GO analysis with up to 10 significantly enriched terms in (a) biological process (BP), (b) cell component (CC) and molecular function (MF), respectively. For 337
Homo’s proteins in the proposed dataset, 6100 biological processes are enriched and 4248 are statistically significant. Single-organism processes are the most important biological process for homo MPs. Six hundred and forty-two (642) cell components are enriched and 401 are statistically significant. Intracellular, cytoplasmic, and organelle are the top three CC enrichments, but they are not much better than others; membrane proteins are distributed in almost all organelles without significant difference. One thousand one hundred and fifty-three (1153) molecular functions are enriched and 561 are statistically significant. Binding with ligands is the most important function of membrane proteins.
KEGG (Kyoto Encyclopedia of Genes and Genomes) is a manually curated pathway database for understanding high-level functions and utilities of the biological system. The KEGG pathway is a collection of manually drawn pathway maps. The enrichment analysis of the KEGG pathway helps researchers understand the pathway a given set of proteins are involved in. For
Homo’s ligand-binding MPs, enriched processes are shown in
Figure 7.
2.9. Performance Evaluation
The proposed prediction model MPLs-Pred was first evaluated by 10-fold cross validation on the training dataset. First, positive samples and negative samples were randomly divided into 10 equal parts, respectively. Then, one part of the positive and negative subset was chosen to build the validation dataset, and the remaining samples were used for training. This process was repeated ten times to build ten sub-predictors. The final performance is the average value of the sub-predictors. Then, independent validation was used to evaluate the generalization of the proposed method. Four metrics were employed to evaluate the performance of the predictor—specificity (Spe), sensitivity (Sen), accuracy (ACC), and the Matthews correlation coefficient (MCC):
where TP, FP, TN, and FN represent true positive, false positive, true negative and false negative, respectively.
The prediction of membrane protein-ligand binding residues is a typical imbalanced learning problem where the number of non-binding residues is significantly more than that of binding residues. Thus, excessive pursuit of overall accuracy is one-sided. In the field of practical application, researchers always expect that the predictor can provide more accuracy of ligand-binding residues instead of non-binding ones in order to obtain more candidate targets. In view of this, the MCC value, which provides an overall measurement of performance of binary classification problems, is regarded as the most reliable evaluation index in the experiments of this paper. The performance of the predictor is positively correlated with the MCC value.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}