Proteomic Analysis of Morphologically Changed Tissues after Prolonged Dexamethasone Treatment

 and
and
Abstract
:
1. Introduction
2. Results
2.1. Clinical Phenotypes
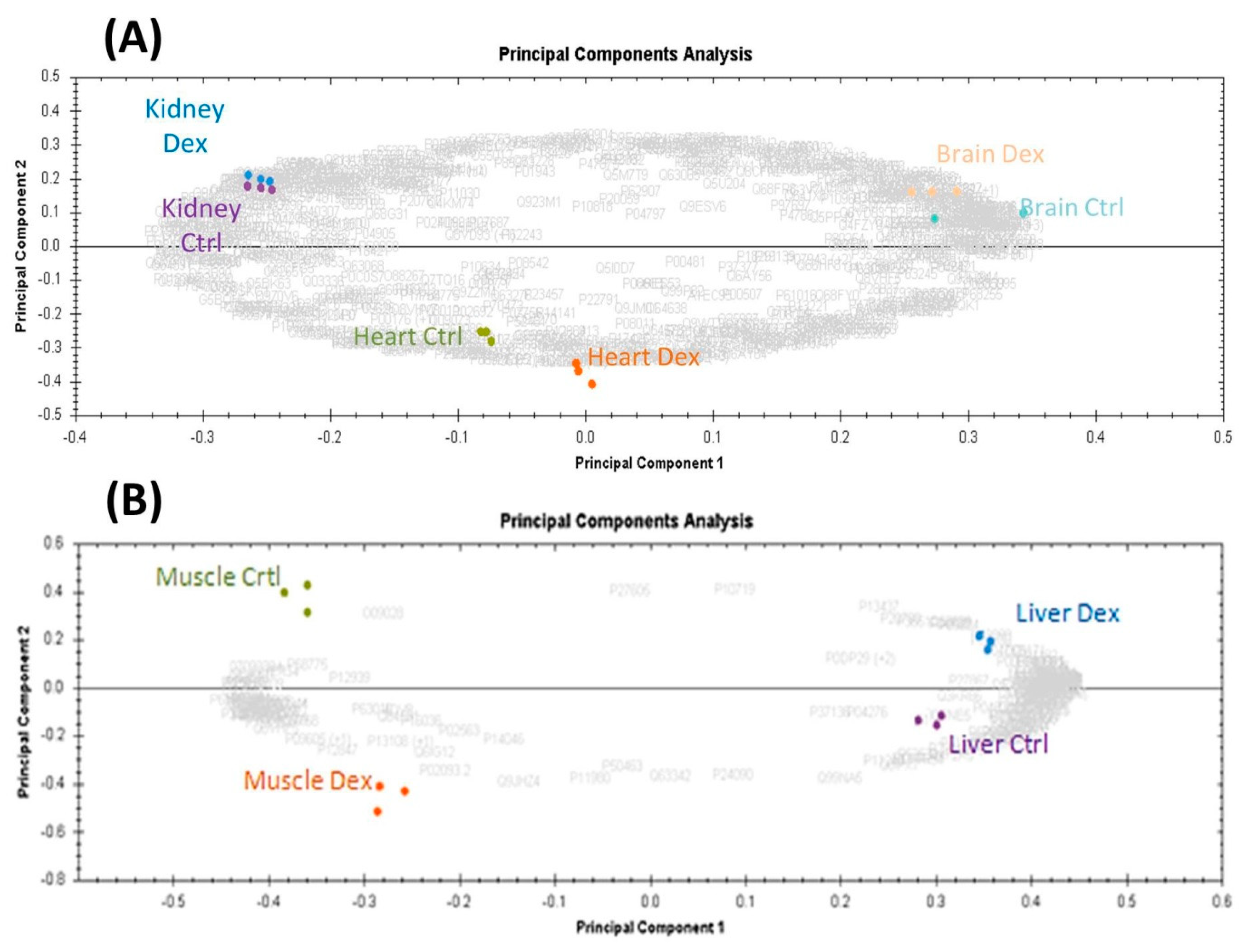
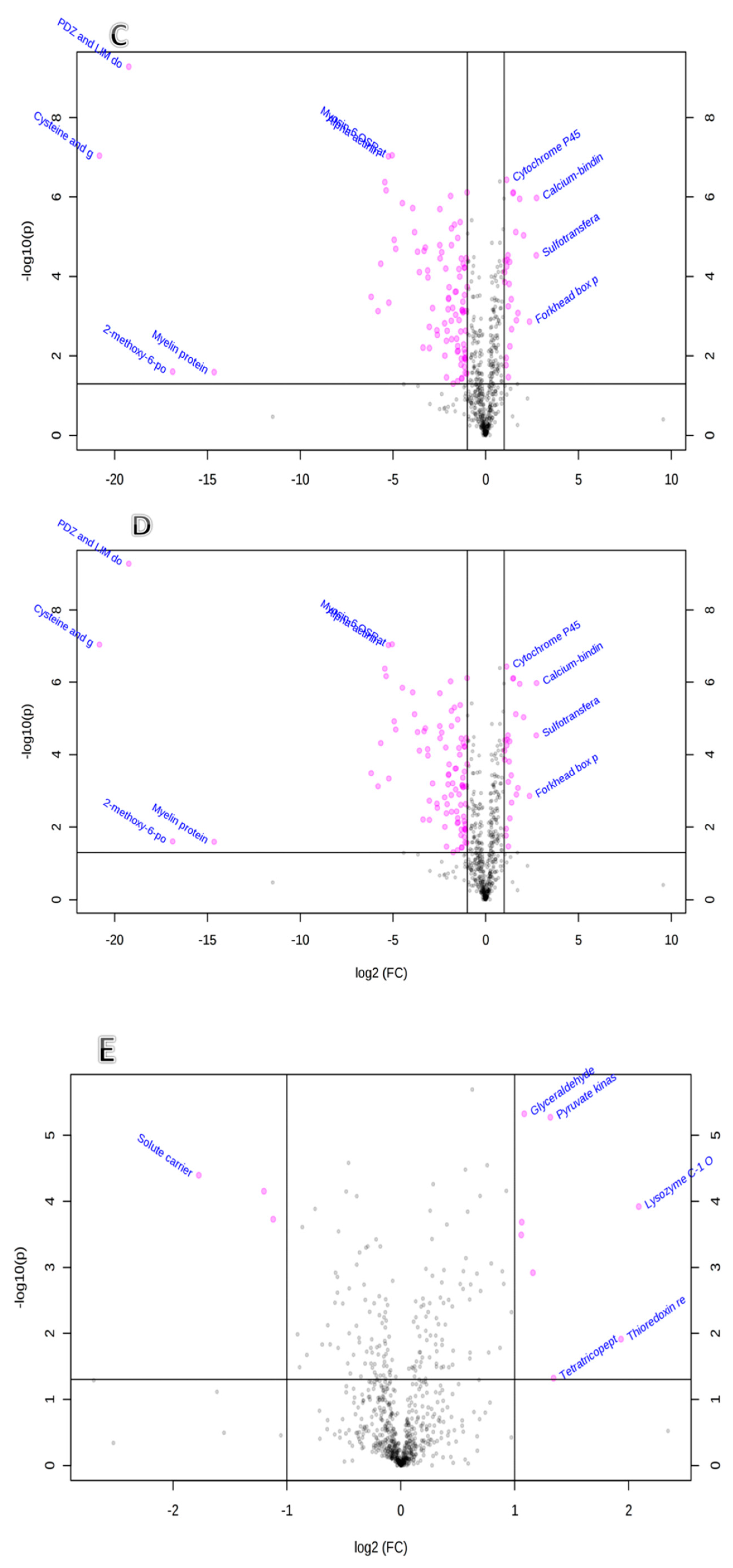
2.2. Mass Spectromeric Protein Identification and Analyses
2.2.1. Gene Ontology and Functional Analysis of the Identified Proteins in the Five Organs
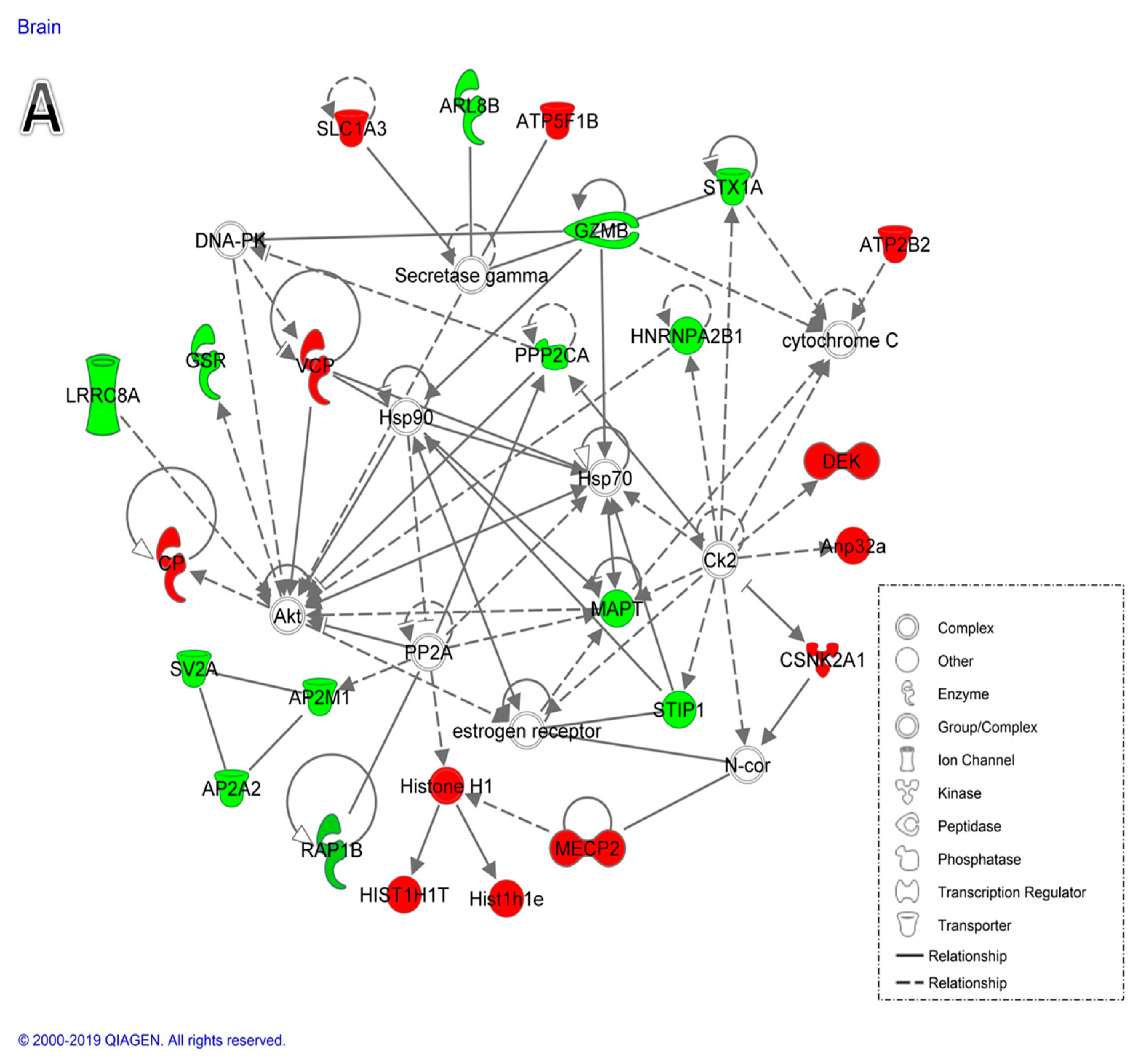
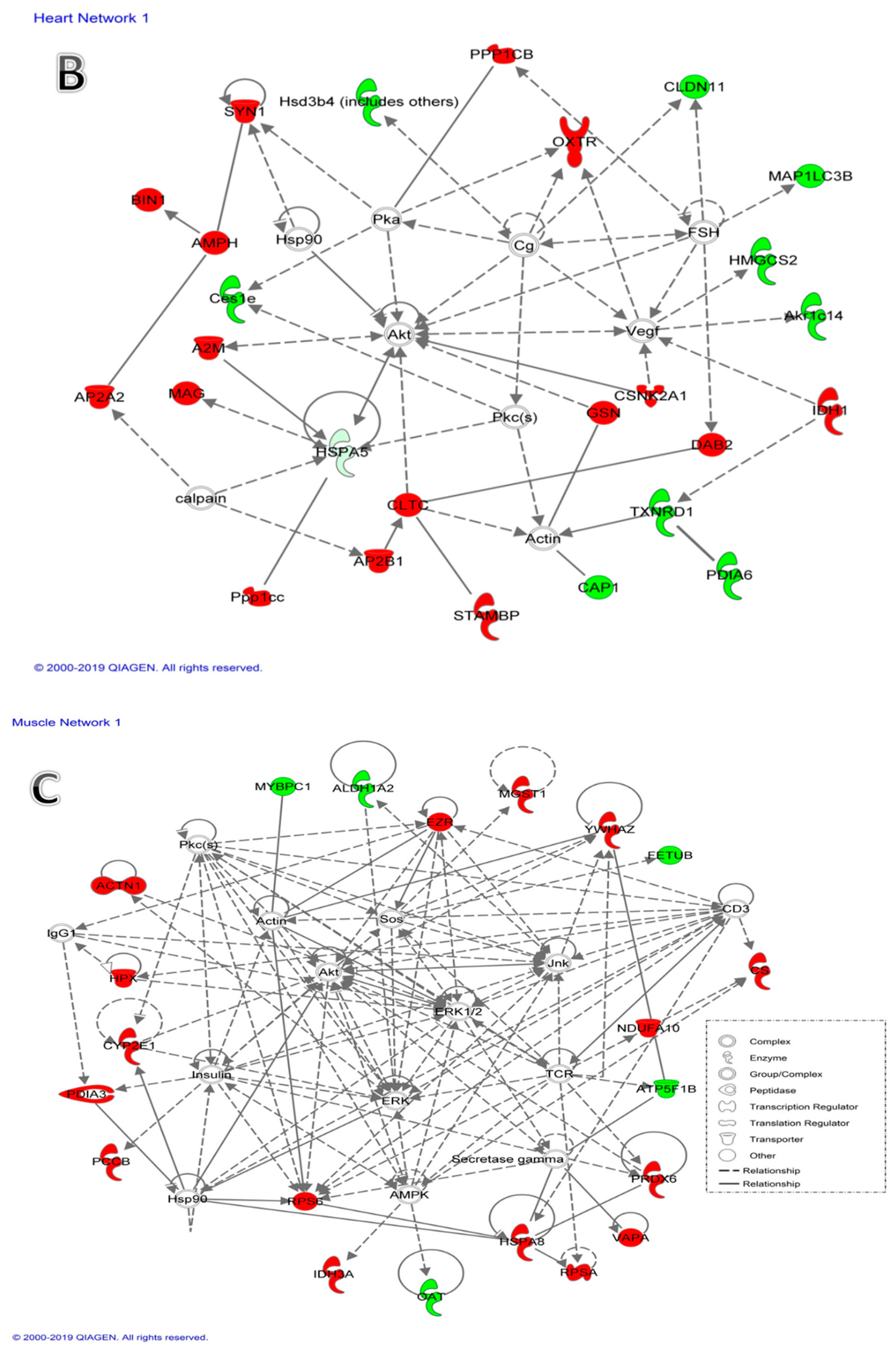
2.2.2. Network and Pathway Analysis
3. Discussion
3.1. Proteomic Profiling of the Major Tissues Affected by Dex Treatment
3.1.1. Proteins Related to the Reduced Brain Size after Dex Treatment
3.1.2. Proteins Related to Decreased Heart Size after Dex Treatment
3.1.3. Proteins Related to Muscle Atrophy after Dex Treatment
3.1.4. Proteins Altered in the Liver After Dex Treatment
3.1.5. Proteins Altered in the Kidney After Dex Treatment
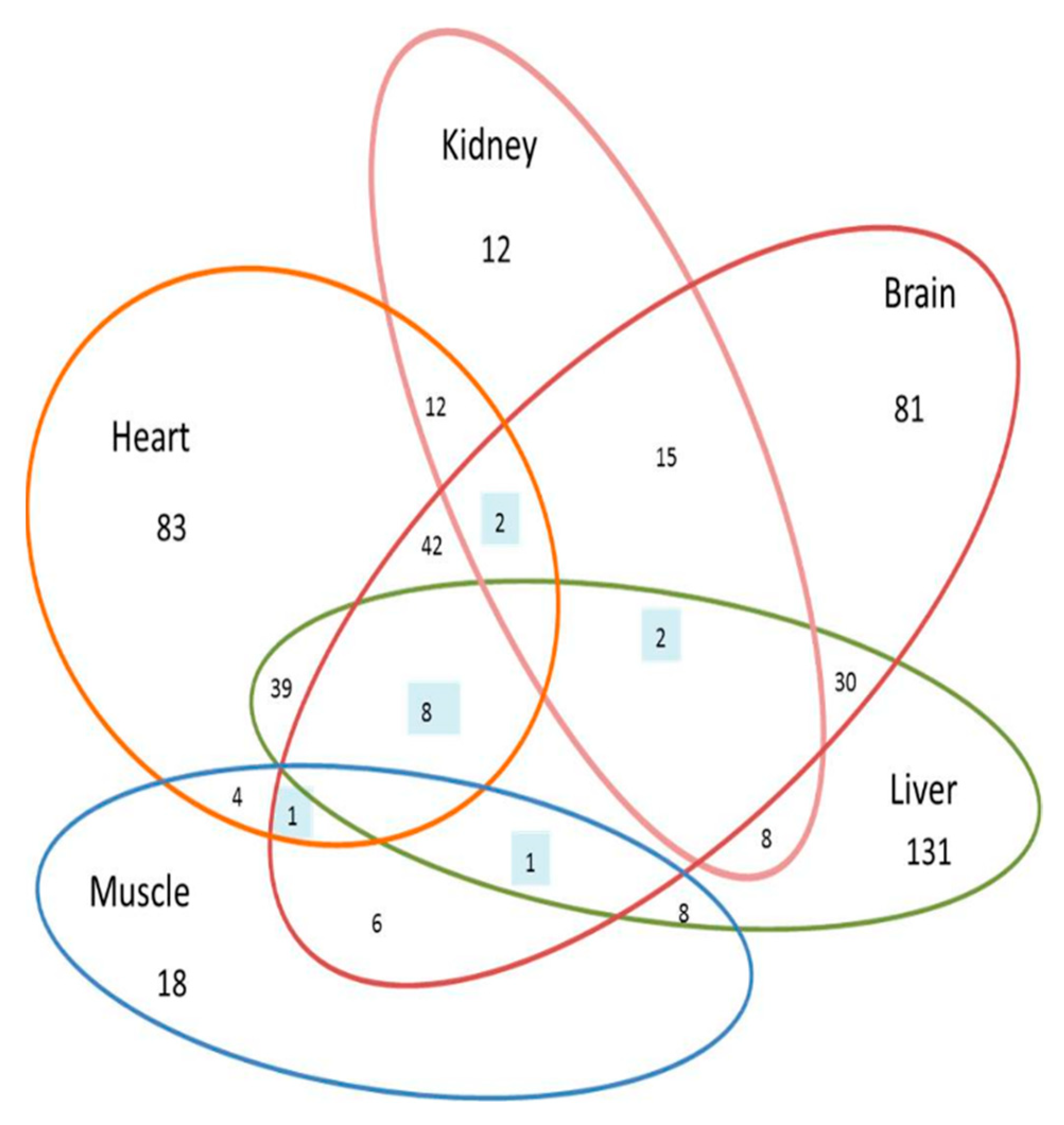
3.1.6. Proteins Altered in All Organs Treated with Dex
3.2. Comparison Amongst the Morhologically Altered Organs after Dex Treatment
4. Materials and Methods
4.1. Ethical Considerations
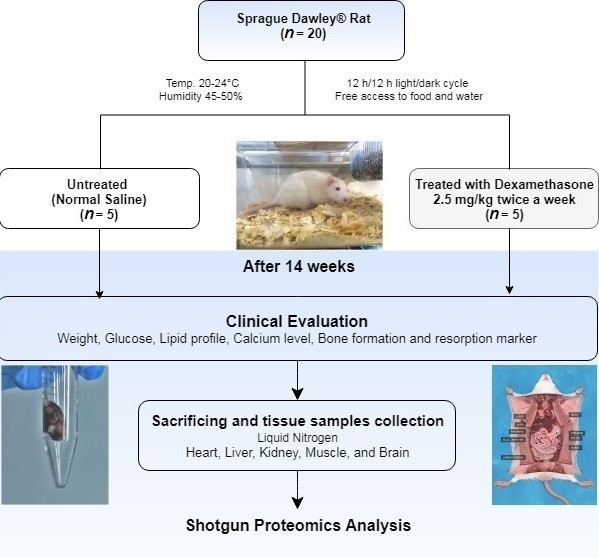
4.2. Experimental Design
4.3. Chemicals and Reagents
4.4. Proteomics
4.4.1. Sample Preparation for Label-Free Protein In-Solution Digestion
4.4.2. Protein Identification by LC-MSE SynaptG2 Platform
4.5. Data Analysis and Informatics
4.6. Bioinformatics Analysis of Proteins in the Tissues and Network Pathway Analysis:
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newton, R. Molecular mechanisms of glucocorticoid action: What is important? Thorax 2000, 55, 603–613. Available online: http://www.ncbi.nlm.nih.gov/pubmed/10856322 (accessed on 15 February 2018). [CrossRef] [PubMed]
- Barnes, P.J. Anti-inflammatory actions of glucocorticoids: Molecular mechanisms. Clin. Sci. (Lond.) 1998, 94, 557–572. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9854452 (accessed on 15 February 2018). [CrossRef]
- Stahn, C.; Buttgereit, F. Genomic and nongenomic effects of glucocorticoids. Nat. Clin. Pract. Rheumatol. 2008, 4, 525–533. [Google Scholar] [CrossRef] [PubMed]
- John, S.; Sabo, P.J.; Thurman, R.E.; Sung, M.H.; Biddie, S.C.; Johnson, T.A.; Hager, G.L.; Stamatoyannopoulos, J.A. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011, 43, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Phuc Le, P.; Friedman, J.R.; Schug, J.; Brestelli, J.E.; Parker, J.B.; Bochkis, I.M.; Kaestner, K.H. Glucocorticoid receptor-dependent gene regulatory networks. PLoS Genet. 2005, 1, e16. [Google Scholar] [CrossRef]
- Yu, C.Y.; Mayba, O.; Lee, J.V.; Tran, J.; Harris, C.; Speed, T.P.; Wang, J.C. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS ONE 2010, 5, e15188. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.; Lew, M.J.; Mayba, O.; Harris, C.A.; Speed, T.P.; Wang, J.C. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 11160–11165. [Google Scholar] [CrossRef] [Green Version]
- Shintani, N.; Hunziker, E.B. Differential effects of dexamethasone on the chondrogenesis of mesenchymal stromal cells: Influence of microenvironment, tissue origin and growth factor. Eur. Cell. Mater. 2011, 22, 302–319; discussion 319–320. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22116649 (accessed on 5 February 2018). [CrossRef]
- McDonough, A.K.; Curtis, J.R.; Saag, K.G. The epidemiology of glucocorticoid-associated adverse events. Curr. Opin. Rheumatol. 2008, 20, 131–137. [Google Scholar] [CrossRef]
- Malkawi, A.K.; Alzoubi, K.H.; Jacob, M.; Matic, G.; Ali, A.; Al Faraj, A.; Almuhanna, F.; Dasouki, M.; Abdel Rahman, A.M.A. Metabolomics based profiling of dexamethasone side effects in rats. Front. Pharmacol. 2018, 9, 46. [Google Scholar] [CrossRef]
- Hohn, A.; Iovino, I.; Cirillo, F.; Drinhaus, H.; Kleinbrahm, K.; Boehm, L.; de Robertis, E.; Hinkelbein, J. Bioinformatical analysis of organ-related (heart, brain, liver, and kidney) and serum proteomic data to identify protein regulation patterns and potential sepsis biomarkers. BioMed. Res. Int. 2018, 2018, 3576157. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, D.; Liu, P.; Yu, Y. Proteomics analysis of water insoluble-urea soluble crystallins from normal and dexamethasone exposed lens. Mol. Vis. 2011, 17, 3423–3436. Available online: http://www.ncbi.nlm.nih.gov/pubmed/22219638 (accessed on 7 August 2018). [PubMed]
- Wang, L.; Zhao, W.C.; Yin, X.L.; Ge, J.Y.; Bu, Z.G.; Ge, H.Y.; Meng, Q.F.; Liu, P. Lens proteomics: Analysis of rat crystallins when lenses are exposed to dexamethasone. Mol. BioSyst. 2012, 8, 888–901. [Google Scholar] [CrossRef] [PubMed]
- Miyara, N.; Shinzato, M.; Yamashiro, Y.; Iwamatsu, A.; Kariya, K.I.; Sawaguchi, S. Proteomic analysis of rat retina in a steroid-induced ocular hypertension model: Potential vulnerability to oxidative stress. Jpn. J. Ophthalmol. 2008, 52, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Celebi, B.; Elcin, A.E.; Elcin, Y.M. Proteome analysis of rat bone marrow mesenchymal stem cell differentiation. J. Proteome Res. 2010, 9, 5217–5227. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, Y.; Takeba, Y.; Kumai, T.; Matsumoto, N.; Mizuno, M.; Murano, K.; Asoh, K.; Takagi, M.; Yamamoto, H.; Kobayashi, S. Antenatal glucocorticoid therapy increase cardiac alpha-enolase levels in fetus and neonate rats. Life Sci. 2009, 85, 609–616. [Google Scholar] [CrossRef]
- Man, W.J.; White, I.R.; Bryant, D.; Bugelski, P.; Camilleri, P.; Cutler, P.; Heald, G.; Lord, P.G.; Wood, J.; Kramer, K. Protein expression analysis of drug-mediated hepatotoxicity in the sprague-dawley rat. Proteomics 2002, 2, 1577–1585. [Google Scholar] [CrossRef]
- Biancotto, G.; Bovo, D.; Mastrorilli, E.; Manuali, E.; Angeletti, R.; Stella, R. Tmt-based proteomics profiling of bovine liver underscores protein markers of anabolic treatments. Proteomics 2019, e1800422. [Google Scholar] [CrossRef]
- Masood, A.; Benabdelkamel, H.; Alfadda, A.A. Obesity proteomics: An update on the strategies and tools employed in the study of human obesity. High-Throughput 2018, 7, 27. [Google Scholar] [CrossRef]
- Hinkelbein, J.; Braunecker, S.; Danz, M.; Bohm, L.; Hohn, A. Time dependent pathway activation of signalling cascades in rat organs after short-term hyperoxia. Int. J. Mol. Sci. 2018, 19, 1960. [Google Scholar] [CrossRef]
- Peptide Atlas. Institute for Systems Biology. 2004–2015. Available online: http://www.peptideatlas.org/PASS/PASS01328 (accessed on 15 February 2018).
- Patel, R.; Patel, M.; Tsai, R.; Lin, V.; Bookout, A.L.; Zhang, Y.; Magomedova, L.; Li, T.; Chan, J.F.; Budd, C.; et al. LXRbeta is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J. Clin. Investig. 2011, 121, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Molecular mechanisms and cellular effects of glucocorticosteroids. Clin. N. Am. 2005, 25, 451–468. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.J.; Blendy, J.A.; Monaghan, A.P.; Krieglstein, K.; Schmid, W.; Aguzzi, A.; Fantuzzi, G.; Hummler, E.; Unsicker, K.; Schutz, G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995, 9, 1608–1621. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7628695 (accessed on 15 February 2018). [CrossRef] [PubMed]
- Chivers, J.E.; Gong, W.; King, E.M.; Seybold, J.; Mak, J.C.; Donnelly, L.E.; Holden, N.S.; Newton, R. Analysis of the dissociated steroid ru24858 does not exclude a role for inducible genes in the anti-inflammatory actions of glucocorticoids. Mol. Pharmacol. 2006, 70, 2084–2095. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.; Eyers, C.E. Analysis of post-translational modifications by lc-ms/ms. Methods Mol. Biol. 2010, 658, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.M.; Rose, S.R. Use of glucocorticoids for the fetus and preterm infant. Clin. Perinatol. 2018, 45, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, R.E., Jr.; Maurer, M.H.; Hunzinger, C.; Lewicka, S.; Buergers, H.F.; Kalenka, A.; Hinkelbein, J.; Broemme, J.O.; Seidler, G.H.; Martin, E.; et al. Reduction in rat phosphatidylethanolamine binding protein-1 (pebp1) after chronic corticosterone treatment may be paralleled by cognitive impairment: A first study. Stress 2008, 11, 134–147. [Google Scholar] [CrossRef]
- Skynner, H.A.; Amos, D.P.; Murray, F.; Salim, K.; Knowles, M.R.; Munoz-Sanjuan, I.; Camargo, L.M.; Bonnert, T.P.; Guest, P.C. Proteomic analysis identifies alterations in cellular morphology and cell death pathways in mouse brain after chronic corticosterone treatment. Brain Res. 2006, 1102, 12–26. [Google Scholar] [CrossRef]
- de Vries, W.B.; van der Leij, F.R.; Bakker, J.M.; Kamphuis, P.J.; van Oosterhout, M.F.; Schipper, M.E.; Smid, G.B.; Bartelds, B.; van Bel, F. Alterations in adult rat heart after neonatal dexamethasone therapy. Pediatr. Res. 2002, 52, 900–906. [Google Scholar] [CrossRef]
- Bentson, J.; Reza, M.; Winter, J.; Wilson, G. Steroids and apparent cerebral atrophy on computed tomography scans. J. Comput. Assist. Tomogr. 1978, 2, 16–23. Available online: http://www.ncbi.nlm.nih.gov/pubmed/670467 (accessed on 17 June 2018). [CrossRef]
- Joshi, Y.B.; Chu, J.; Pratico, D. Stress hormone leads to memory deficits and altered tau phosphorylation in a model of alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.; Vaz-Silva, J.; Pinto, V.; Dalla, C.; Kokras, N.; Bedenk, B.; Mack, N.; Czisch, M.; Almeida, O.F.; Sousa, N.; et al. Tau protein is essential for stress-induced brain pathology. Proc. Natl. Acad. Sci. USA 2016, 113, E3755–E3763. [Google Scholar] [CrossRef] [PubMed]
- Haynes, L.E.; Griffiths, M.R.; Hyde, R.E.; Barber, D.J.; Mitchell, I.J. Dexamethasone induces limited apoptosis and extensive sublethal damage to specific subregions of the striatum and hippocampus: Implications for mood disorders. Neuroscience 2001, 104, 57–69. Available online: http://www.ncbi.nlm.nih.gov/pubmed/11311531 (accessed on 17 June 2018). [CrossRef]
- Rassoulpour, A.; Wu, H.Q.; Ferre, S.; Schwarcz, R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J. Neurochem. 2005, 93, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Cidlowski, J.A. Glucocorticoid signaling in the heart: A cardiomyocyte perspective. J. Steroid Biochem. Mol. Biol. 2015, 153, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Oakley, R.H.; Cruz-Topete, D.; Cidlowski, J.A. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology 2012, 153, 5346–5360. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.H.; Im, N.N.; Huilgol, S.V.; Yendigeri, S.M.; Narendar, K.; Rajasekhar, C.H. Dose dependent hepatic and endothelial changes in rats treated with dexamethasone. J. Clin. Diagn. Res. 2015, 9, FF08–FF10. [Google Scholar] [CrossRef]
- Seckl, J.R.; Walker, B.R. Minireview: 11beta-hydroxysteroid dehydrogenase type 1- a tissue-specific amplifier of glucocorticoid action. Endocrinology 2001, 142, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Redman, R.A.; Logg, C.R.; Coetzee, G.A.; Kasahara, N.; Frenkel, B. Glucocorticoids inhibit developmental stage-specific osteoblast cell cycle. Dissociation of cyclin a-cyclin-dependent kinase 2 from e2f4-p130 complexes. J. Biol. Chem. 2000, 275, 19992–20001. [Google Scholar] [CrossRef]
- Porrello, E.R.; Meeker, W.F.; Thomas, W.G.; Widdop, R.E.; Delbridge, L.M. Glucocorticoids suppress growth in neonatal cardiomyocytes co-expressing at(2) and at(1) angiotensin receptors. Neonatology 2010, 97, 257–265. [Google Scholar] [CrossRef]
- Shimizu, K.; Genma, R.; Gotou, Y.; Nagasaka, S.; Honda, H. Three-dimensional culture model of skeletal muscle tissue with atrophy induced by dexamethasone. Bioengineering (Basel) 2017, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- De Vos, P.; Saladin, R.; Auwerx, J.; Staels, B. Induction of ob gene expression by corticosteroids is accompanied by body weight loss and reduced food intake. J. Biol. Chem. 1995, 270, 15958–15961. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7608151 (accessed on 15 May 2019). [CrossRef] [PubMed]
- Konagaya, M.; Bernard, P.A.; Max, S.R. Blockade of glucocorticoid receptor binding and inhibition of dexamethasone-induced muscle atrophy in the rat by ru38486, a potent glucocorticoid antagonist. Endocrinology 1986, 119, 375–380. [Google Scholar] [CrossRef]
- Singleton, J.R.; Baker, B.L.; Thorburn, A. Dexamethasone inhibits insulin-like growth factor signaling and potentiates myoblast apoptosis. Endocrinology 2000, 141, 2945–2950. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Nader, G.A. Balancing muscle hypertrophy and atrophy. Nat. Med. 2004, 10, 584–585. [Google Scholar] [CrossRef]
- Hwang, S.L.; Jeong, Y.T.; Li, X.; Kim, Y.D.; Lu, Y.; Chang, Y.C.; Lee, I.K.; Chang, H.W. Inhibitory cross-talk between the AMPK and ERK pathways mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Br. J. Pharmacol. 2013, 169, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Tan, K.; Carey, K.T.; Fukushima, A.; Tiganis, T.; Cole, T.J. Glucocorticoids stimulate hepatic and renal catecholamine inactivation by direct rapid induction of the dopamine sulfotransferase sult1d1. Endocrinology 2010, 151, 185–194. [Google Scholar] [CrossRef]
- Yin, G.; Cao, L.; Du, J.; Jia, R.; Kitazawa, T.; Kubota, A.; Teraoka, H. Dexamethasone-induced hepatomegaly and steatosis in larval zebrafish. J. Toxicol. Sci. 2017, 42, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Park, S.; Cheon, B.; Kim, H.W.; Yu, D.; Choi, J. Body weight, blood pressure, and systemic changes following low-dosage prednisolone administration in dogs. Am. J. Vet. Res. 2017, 78, 1091–1097. [Google Scholar] [CrossRef]
- Ayyar, V.S.; Almon, R.R.; DuBois, D.C.; Sukumaran, S.; Qu, J.; Jusko, W.J. Functional proteomic analysis of corticosteroid pharmacodynamics in rat liver: Relationship to hepatic stress, signaling, energy regulation, and drug metabolism. J. Proteom. 2017, 160, 84–105. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Li, Y.; He, L.; Agarwal, A.R.; Zeng, N.; Cadenas, E.; Stiles, B.L. PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free Radic. Biol. Med. 2013, 60, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordag, N.; Klie, S.; Jurchott, K.; Vierheller, J.; Schiewe, H.; Albrecht, V.; Tonn, J.C.; Schwartz, C.; Schichor, C.; Selbig, J. Glucocorticoid (dexamethasone)-induced metabolome changes in healthy males suggest prediction of response and side effects. Sci. Rep. 2015, 5, 15954. [Google Scholar] [CrossRef] [PubMed]
- Baylis, C.; Handa, R.K.; Sorkin, M. Glucocorticoids and control of glomerular filtration rate. Semin. Nephrol. 1990, 10, 320–329. Available online: http://www.ncbi.nlm.nih.gov/pubmed/2200095 (accessed on 2 September 2018). [PubMed]
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2011, 163, 29–43. [Google Scholar] [CrossRef] [PubMed]
- de Haij, S.; Daha, M.R.; van Kooten, C. Mechanism of steroid action in renal epithelial cells. Kidney Int. 2004, 65, 1577–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Zoorob, R.J.; Cender, D. A different look at corticosteroids. Am. Fam. Physician 1998, 58, 443–450. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9713398 (accessed on 15 February 2018). [PubMed]
- Acevedo, A.; Berthel, A.; DuBois, D.; Almon, R.R.; Jusko, W.J.; Androulakis, I.P. Pathway-based analysis of the liver response to intravenous methylprednisolone administration in rats: Acute versus chronic dosing. Gene Regul. Syst. Biol. 2019, 13, 1177625019840282. [Google Scholar] [CrossRef]
- Li, Y.; Yang, W. Preventive effects of nitroglycerine on glucocorticoid-induced osteoporosis in growing rats. J. Huazhong Univ. Sci. Technol. Med.Sci. 2007, 27, 528–531. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, X.; Zhao, L.; Li, F.; Xiong, Z. Kidney tissue targeted metabolic profiling of glucocorticoid-induced osteoporosis and the proposed therapeutic effects of rhizoma drynariae studied using UHPLC/MS/MS. Biomed. Chromatogr. 2014, 28, 878–884. [Google Scholar] [CrossRef]
- Alaiya, A.; Fox, J.; Bobis, S.; Matic, G.; Shinwari, Z.; Barhoush, E.; Marquez, M.; Nilsson, S.; Holmberg, A.R. Proteomic analysis of soft tissue tumor implants treated with a novel polybisphosphonate. Cancer Genom. Proteom. 2014, 11, 39–49. Available online: http://www.ncbi.nlm.nih.gov/pubmed/24633318 (accessed on 15 April 2019).
- UniProt. Knowledgebase 2002–2019. Available online: https://www.uniprot.org/ (accessed on 15 February 2018).
- Li, G.Z.; Vissers, J.P.; Silva, J.C.; Golick, D.; Gorenstein, M.V.; Geromanos, S.J. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 2009, 9, 1696–1719. [Google Scholar] [CrossRef] [PubMed]
- Alaiya, A.A.; Aljurf, M.; Shinwari, Z.; Almohareb, F.; Malhan, H.; Alzahrani, H.; Owaidah, T.; Fox, J.; Alsharif, F.; Mohamed, S.Y.; et al. Protein signatures as potential surrogate biomarkers for stratification and prediction of treatment response in chronic myeloid leukemia patients. Int. J. Oncol. 2016, 49, 913–933. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Web-based inference of biological patterns, functions and pathways from metabolomic data using metaboanalyst. Nat. Protoc. 2011, 6, 743–760. [Google Scholar] [CrossRef] [PubMed]
- QIAGEN Bioinformatics. Ingenuity Pathway Analysis. Available online: https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis (accessed on 15 February 2018).
- Long, F.; Liu, H.; Hahn, C.; Sumazin, P.; Zhang, M.Q.; Zilberstein, A. Genome-wide prediction and analysis of function-specific transcription factor binding sites. In Silico Biol. 2004, 4, 395–410. Available online: http://www.ncbi.nlm.nih.gov/pubmed/15506990 (accessed on 5 December 2018). [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heart | Muscle | Liver | Kidney | |||||
|---|---|---|---|---|---|---|---|---|
| Brain | Acot1+ ACOT7 Aldh1a7 AP2A2 AP2M1 ATP2B2 ATP5PF++ ATP6V0A1 CEP83 CES1 CHDH† CLTRN CPS1+ CSNK2A1 | Cyp2d1/Cyp2d5+ DBI ECHDC3 EIF2D EPHX1 GSTZ1+ HEATR6 IDH1 MPC2++ Mug1+ MYL2+ NENF PAFAH1B2 CTSC | PDIA6 PNP PYGL+ RAB7A RALA RNLS RPS2 RPS16+ STAMBP STIP1 STX1A SYN2 Wasl | ATP5F1B CHDH† HPX MGST1 PRDX6 RIPOR2‡‡ | Acot1 AKR1A1 AKR1B1 BCKDHA CA1 COX5B CPS1 Cyp2d1/Cyp2d5 GAPDH†† GCSH GDI1 GSTZ1 HADH HMGCS1 KHK | LDHB LRRC8 CLYPLA1 Mug1 MYH6 MYL2 PHYHIPL PYGL RIPOR2‡‡ RPS16 SLC25A4 SLC25A5 SUOX Tpm2 VPS45 | ATP5PF++ CAPZB CORO6 COTL1 CSRP1 GAPDH†† GRN MCCC1 MPC2++ MYH6 NCALD RAB3A RAB3C SCARB1 UCMA | |
| Heart | CHDH EZR NDUFA10 RPSA | Acot1+ ACOX1 Akr1c14 ALDOB AMACR ASS1 ATP5PO BAAT BHMT2 BIN1 Ces1e CPS1+ | CSRP3 Cyp2d1/Cyp2d5+ EEF1A2 FABP1 FABP3 FBP1 FHL2 Gnmt Gsta1 GSTA3 GSTM2 GSTZ1+ | HMGCS2 HSPA5 MAT1A Mug1+ MYL2+ NDUFA9 PYGL+ RPS16+ Sult1a1 TKFC UGT2B15 UPB1 | ALDH1B1 ATP5PF BGN CDKL3 FABP1‡ GSTM1 GSTM2‡ Hsd3b4 MPC2 THNSL2 TXNRD1 Vamp1 | |||
| Liver | ABHD17B ACD ACTN1 CS IDH3A KRT24 PC RIPOR2‡‡ | AHSG FABP1‡ GSTM2‡ PDHB PKM Tpm3 GAPDHS MYH6 | ||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malkawi, A.K.; Masood, A.; Shinwari, Z.; Jacob, M.; Benabdelkamel, H.; Matic, G.; Almuhanna, F.; Dasouki, M.; Alaiya, A.A.; Rahman, A.M.A. Proteomic Analysis of Morphologically Changed Tissues after Prolonged Dexamethasone Treatment. Int. J. Mol. Sci. 2019, 20, 3122. https://doi.org/10.3390/ijms20133122
Malkawi AK, Masood A, Shinwari Z, Jacob M, Benabdelkamel H, Matic G, Almuhanna F, Dasouki M, Alaiya AA, Rahman AMA. Proteomic Analysis of Morphologically Changed Tissues after Prolonged Dexamethasone Treatment. International Journal of Molecular Sciences. 2019; 20(13):3122. https://doi.org/10.3390/ijms20133122
Chicago/Turabian StyleMalkawi, Abeer K., Afshan Masood, Zakia Shinwari, Minnie Jacob, Hicham Benabdelkamel, Goran Matic, Falah Almuhanna, Majed Dasouki, Ayodele A. Alaiya, and Anas M. Abdel Rahman. 2019. "Proteomic Analysis of Morphologically Changed Tissues after Prolonged Dexamethasone Treatment" International Journal of Molecular Sciences 20, no. 13: 3122. https://doi.org/10.3390/ijms20133122
APA StyleMalkawi, A. K., Masood, A., Shinwari, Z., Jacob, M., Benabdelkamel, H., Matic, G., Almuhanna, F., Dasouki, M., Alaiya, A. A., & Rahman, A. M. A. (2019). Proteomic Analysis of Morphologically Changed Tissues after Prolonged Dexamethasone Treatment. International Journal of Molecular Sciences, 20(13), 3122. https://doi.org/10.3390/ijms20133122