Autocrine Production of PDGF Stimulated by the Tenascin-C-Derived Peptide TNIIIA2 Induces Hyper-Proliferation in Glioblastoma Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
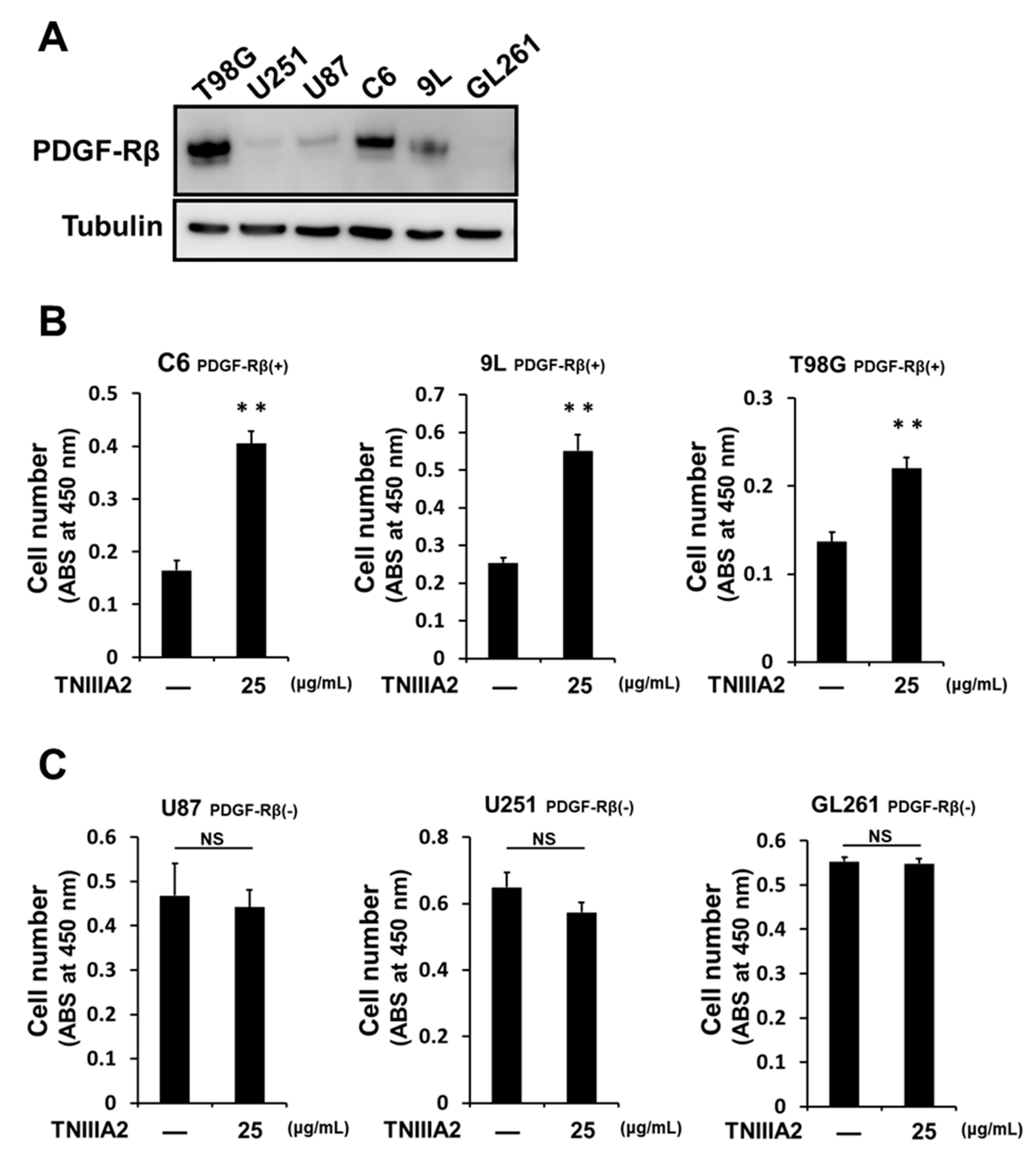
2.1. TNIIIA2 Stimulated GBM Cell Proliferation in an Autocrine/Paracrine Manner through the Induction of PDGF Expression
2.2. TNIIIA2 Enhanced GBM Migration via β1-Integrin/PKCα Pathway
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Cell Proliferation
4.4. Western Blotting
4.5. Wound Healing Assay
4.6. Semiquantitative PCR
4.7. Gelatin Zymography
4.8. Isolation of Rat Brain-Derived Fibroblasts
4.9. Scattering Assay
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PDGF | platelet-derived growth factor |
| GBM | glioblastoma multiforme |
| ECM | extracellular matrix |
| PDGF-R | platelet-derived growth factor receptor |
| PKC | protein kinase C |
| RPMI | Roswell Park Memorial Institute |
| FBS | fetal bovine serum |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| PCR | polymerase chain reaction |
| TBE | tris-borate-EDTA |
| EDTA | ethylenediaminetetraacetic acid |
| EtBr | ethidium bromide |
| MMP | matrix metalloproteinase |
| sup. | Supernatant |
| WCL | whole cell lysates |
References
- Midwood, K.S.; Hussenet, T.; Langlois, B.; Orend, G. Advances in tenascin-C biology. Cell. Mol. Life Sci. CMLS 2011, 68, 3175–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, A.; Yoshida, T.; Tamaki, H.; Sakakura, T. Tenascin expression in cancer cells and stroma of human breast cancer and its prognostic significance. Clin. Cancer Res. 1995, 1, 1035–1041. [Google Scholar] [PubMed]
- Yang, Z.; Ni, W.; Cui, C.; Fang, L.; Xuan, Y. Tenascin C is a prognostic determinant and potential cancer-associated fibroblasts marker for breast ductal carcinoma. Exp. Mol. Pathol. 2017, 102, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Naba, A.; Bhutkar, A.; Guardia, T.; Miller, K.M.; Li, C.M.-C.; Dayton, T.L.; Sanchez-Rivera, F.J.; Kim-Kiselak, C.; Jailkhani, N.; et al. Quantitative proteomics identify Tenascin-C as a promoter of lung cancer progression and contributor to a signature prognostic of patient survival. Proc. Natl. Acad. Sci. USA 2017, 114, E5625–E5634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adhes. Migr. 2014, 9, 48–82. [Google Scholar] [CrossRef] [Green Version]
- Brösicke, N.; van Landeghem, F.K.H.; Scheffler, B.; Faissner, A. Tenascin-C is expressed by human glioma in vivo and shows a strong association with tumor blood vessels. Cell Tissue Res. 2013, 354, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Dueck, M.; Riedl, S.; Hinz, U.; Tandara, A.; Möller, P.; Herfarth, C.; Faissner, A. Detection of tenascin-C isoforms in colorectal mucosa, ulcerative colitis, carcinomas and liver metastases. Int. J. Cancer 1999, 82, 477–483. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef]
- Saito, Y.; Imazeki, H.; Miura, S.; Yoshimura, T.; Okutsu, H.; Harada, Y.; Ohwaki, T.; Nagao, O.; Kamiya, S.; Hayashi, R.; et al. A peptide derived from tenascin-C induces beta1 integrin activation through syndecan-4. J. Biol. Chem. 2007, 282, 34929–34937. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Seki, Y.; Saito, Y.; Kamiya, S.; Fujita, M.; Okutsu, H.; Iyoda, T.; Takai, T.; Owaki, T.; Yajima, H.; et al. Tenascin-C-derived peptide TNIIIA2 highly enhances cell survival and platelet-derived growth factor (PDGF)-dependent cell proliferation through potentiated and sustained activation of integrin α5β1. J. Biol. Chem. 2014, 289, 17699–17708. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Yamamoto, T.; Iyoda, T.; Fujisawa, T.; Sasada, M.; Nagai, R.; Kudo, C.; Otsuka, K.; Kamiya, S.; Kodama, H.; et al. Aggressive Progression in Glioblastoma Cells through Potentiated Activation of Integrin α5β1 by the Tenascin-C-derived Peptide TNIIIA2. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.; Hede, S.-M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF receptors in glioma. Ups. J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nistér, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Brennan, C.; Momota, H.; Hambardzumyan, D.; Ozawa, T.; Tandon, A.; Pedraza, A.; Holland, E. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS ONE 2009, 4, e7752. [Google Scholar] [CrossRef]
- Ozawa, T.; Riester, M.; Cheng, Y.-K.; Huse, J.T.; Squatrito, M.; Helmy, K.; Charles, N.; Michor, F.; Holland, E.C. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell 2014, 26, 288–300. [Google Scholar] [CrossRef]
- Kato, R.; Ishikawa, T.; Kamiya, S.; Oguma, F.; Ueki, M.; Goto, S.; Nakamura, H.; Katayama, T.; Fukai, F. A new type of antimetastatic peptide derived from fibronectin. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2002, 8, 2455–2462. [Google Scholar]
- Siri, A.; Knäuper, V.; Veirana, N.; Caocci, F.; Murphy, G.; Zardi, L. Different Susceptibility of Small and Large Human Tenascin-C Isoforms to Degradation by Matrix Metalloproteinases. J. Biol. Chem. 1995, 270, 8650–8654. [Google Scholar] [CrossRef] [Green Version]
- Elfenbein, A.; Simons, M. Syndecan-4 signaling at a glance. J. Cell Sci. 2013, 126, 3799–3804. [Google Scholar] [CrossRef] [Green Version]
- Lokker, N.A.; Sullivan, C.M.; Hollenbach, S.J.; Israel, M.A.; Giese, N.A. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: Evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002, 62, 3729–3735. [Google Scholar] [PubMed]
- Brösicke, N.; Faissner, A. Role of tenascins in the ECM of gliomas. Cell Adhes. Migr. 2015, 9, 131–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passlick, B.; Sienel, W.; Seen-Hibler, R.; Wöckel, W.; Thetter, O.; Mutschler, W.; Pantel, K. Overexpression of matrix metalloproteinase 2 predicts unfavorable outcome in early-stage non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 3944–3948. [Google Scholar]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef]
- Davis, G.E.; Bayless, K.J.; Davis, M.J.; Meininger, G.A. Regulation of Tissue Injury Responses by the Exposure of Matricryptic Sites within Extracellular Matrix Molecules. Am. J. Pathol. 2000, 156, 1489–1498. [Google Scholar] [CrossRef] [Green Version]
- Carbonell, W.S.; DeLay, M.; Jahangiri, A.; Park, C.C.; Aghi, M.K. β1 integrin targeting potentiates antiangiogenic therapy and inhibits the growth of bevacizumab-resistant glioblastoma. Cancer Res. 2013, 73, 3145–3154. [Google Scholar] [CrossRef]
- Blandin, A.-F.; Renner, G.; Lehmann, M.; Lelong-Rebel, I.; Martin, S.; Dontenwill, M. β1 Integrins as Therapeutic Targets to Disrupt Hallmarks of Cancer. Front. Pharmacol. 2015, 6, 279. [Google Scholar] [CrossRef]
- Jahangiri, A.; Aghi, M.K.; Carbonell, W.S. β1 integrin: Critical path to antiangiogenic therapy resistance and beyond. Cancer Res. 2014, 74, 3–7. [Google Scholar] [CrossRef]
- Janouskova, H.; Maglott, A.; Leger, D.Y.; Bossert, C.; Noulet, F.; Guerin, E.; Guenot, D.; Pinel, S.; Chastagner, P.; Plenat, F.; et al. Integrin α5β1 plays a critical role in resistance to temozolomide by interfering with the p53 pathway in high-grade glioma. Cancer Res. 2012, 72, 3463–3470. [Google Scholar] [CrossRef]
- Matsunaga, T.; Fukai, F.; Miura, S.; Nakane, Y.; Owaki, T.; Kodama, H.; Tanaka, M.; Nagaya, T.; Takimoto, R.; Takayama, T.; et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia 2008, 22, 353–360. [Google Scholar] [CrossRef]
- Iyoda, T.; Nagamine, Y.; Nakane, Y.; Tokita, Y.; Akari, S.; Otsuka, K.; Fujita, M.; Itagaki, K.; Takizawa, Y.-I.; Orita, H.; et al. Coadministration of the FNIII14 Peptide Synergistically Augments the Anti-Cancer Activity of Chemotherapeutic Drugs by Activating Pro-Apoptotic Bim. PLoS ONE 2016, 11, e0162525. [Google Scholar] [CrossRef]
- Oh, E.S.; Woods, A.; Couchman, J.R. Syndecan-4 proteoglycan regulates the distribution and activity of protein kinase C. J. Biol. Chem. 1997, 272, 8133–8136. [Google Scholar] [CrossRef]
- do Carmo, A.; Balça-Silva, J.; Matias, D.; Lopes, M.C. PKC signaling in glioblastoma. Cancer Biol. Ther. 2013, 14, 287–294. [Google Scholar] [CrossRef] [Green Version]
- Amos, S.; Mut, M.; diPierro, C.G.; Carpenter, J.E.; Xiao, A.; Kohutek, Z.A.; Redpath, G.T.; Zhao, Y.; Wang, J.; Shaffrey, M.E.; et al. Protein kinase C-alpha-mediated regulation of low-density lipoprotein receptor related protein and urokinase increases astrocytoma invasion. Cancer Res. 2007, 67, 10241–10251. [Google Scholar] [CrossRef]
- Miekka, S.I.; Ingham, K.C.; Menache, D. Rapid methods for isolation of human plasma fibronectin. Thromb. Res. 1982, 27, 1–14. [Google Scholar] [CrossRef]
- Saginati, M.; Siri, A.; Balza, E.; Ponassi, M.; Zardi, L. A simple procedure for tenascin purification. Eur. J. Biochem. 1992, 205, 545–549. [Google Scholar] [CrossRef]
- Toth, M.; Sohail, A.; Fridman, R. Assessment of Gelatinases (MMP-2 and MMP-9) by Gelatin Zymography. In Metastasis Research Protocols; Dwek, M., Brooks, S.A., Schumacher, U., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 121–135. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujita, M.; Yamamoto, T.; Iyoda, T.; Fujisawa, T.; Nagai, R.; Kudo, C.; Sasada, M.; Kodama, H.; Fukai, F. Autocrine Production of PDGF Stimulated by the Tenascin-C-Derived Peptide TNIIIA2 Induces Hyper-Proliferation in Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 3183. https://doi.org/10.3390/ijms20133183
Fujita M, Yamamoto T, Iyoda T, Fujisawa T, Nagai R, Kudo C, Sasada M, Kodama H, Fukai F. Autocrine Production of PDGF Stimulated by the Tenascin-C-Derived Peptide TNIIIA2 Induces Hyper-Proliferation in Glioblastoma Cells. International Journal of Molecular Sciences. 2019; 20(13):3183. https://doi.org/10.3390/ijms20133183
Chicago/Turabian StyleFujita, Motomichi, Tetsuya Yamamoto, Takuya Iyoda, Tatsuya Fujisawa, Reo Nagai, Chikako Kudo, Manabu Sasada, Hiroaki Kodama, and Fumio Fukai. 2019. "Autocrine Production of PDGF Stimulated by the Tenascin-C-Derived Peptide TNIIIA2 Induces Hyper-Proliferation in Glioblastoma Cells" International Journal of Molecular Sciences 20, no. 13: 3183. https://doi.org/10.3390/ijms20133183