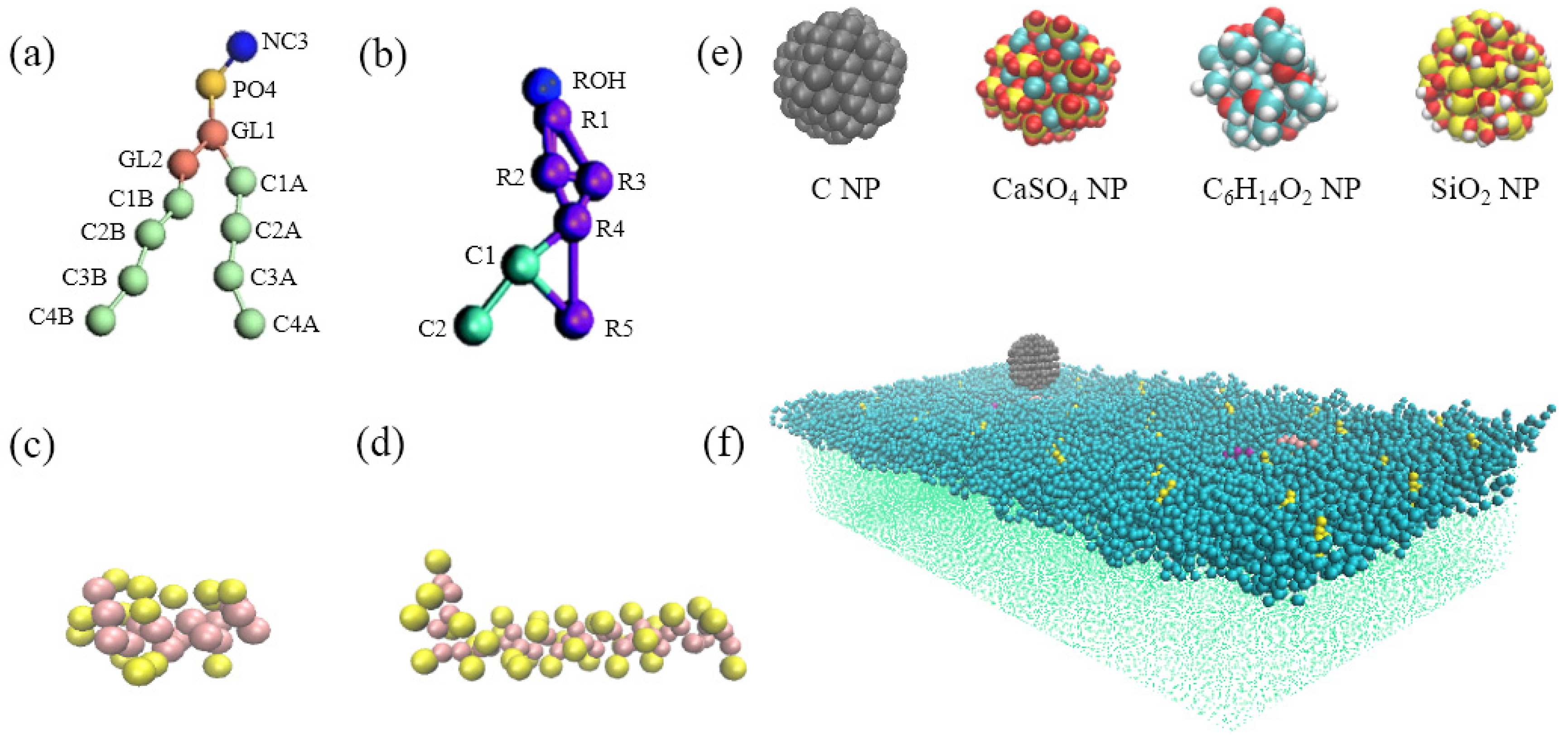
To investigate the interaction mechanism of four kinds of representative NPs with the pulmonary surfactant monolayer during natural breathing patterns, we analyzed the mean-square displacement (MSD) of the NPs, centroid-to-centroid distance between the NP and monolayer, order parameter of the phospholipid molecule, and surface area per lipid molecule in the interaction. The MSD is used to evaluate the movement of a given particle, indicating its diffusion rate [
23]. It is calculated with the following formula:
where
is used to represent the position of a particle at time
t.
2.1. Interactions of Different Nanoparticles (NPs) with the Pulmonary Surfactant Monolayer in Exhalation and Inhalation Breathing States
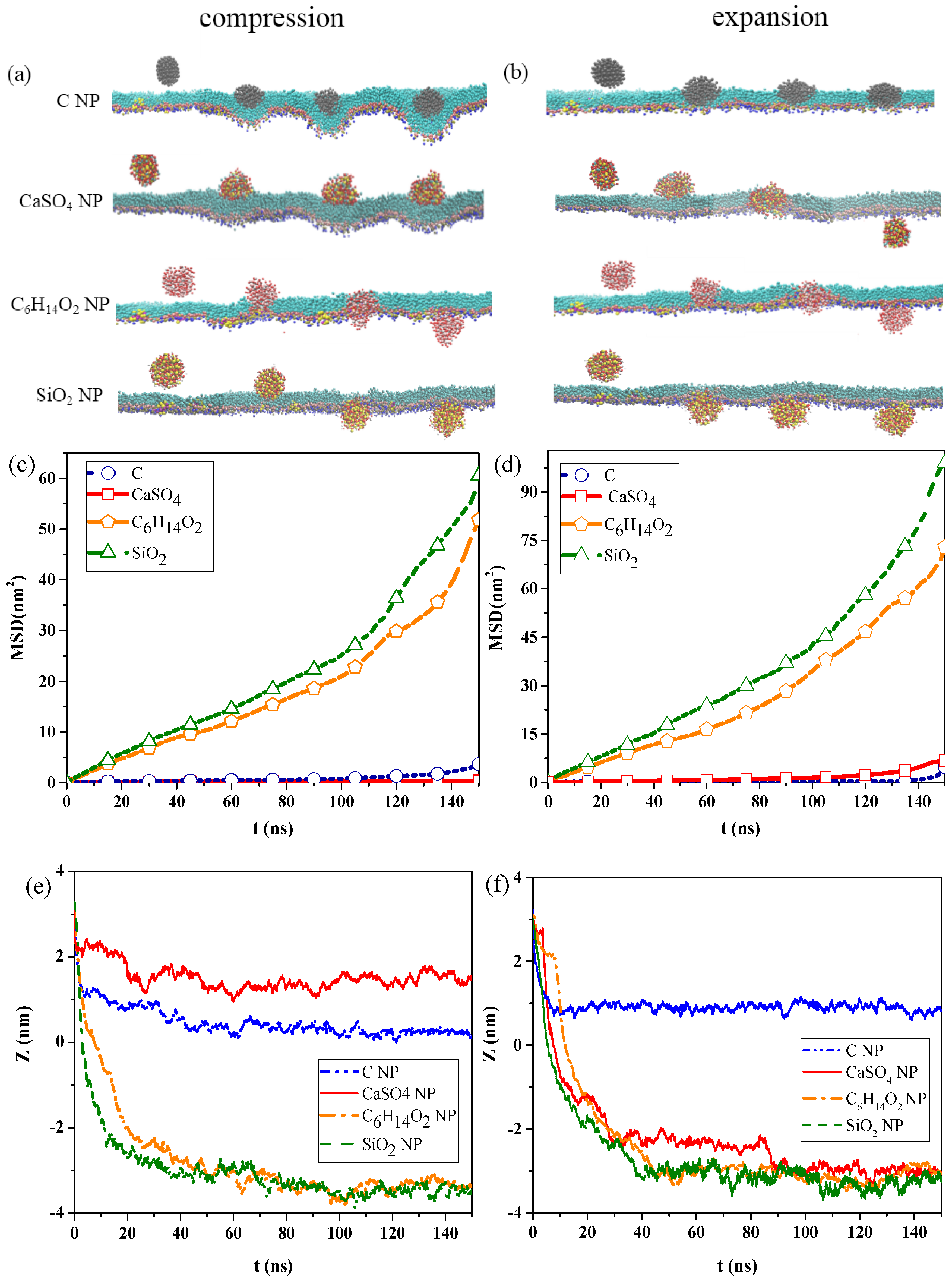
Figure 1a,b show the snapshots of interactions among the different NPs and the pulmonary surfactant monolayer during compression and expansion, respectively. Water molecules are not shown in the figures to emphasize NP movement. We can observe that C and CaSO
4 NPs were embedded into the monolayer at the exhalation breathing state, whereas C
6H
14O
2 and SiO
2 NPs penetrated the monolayer. Compared with CaSO
4, C was almost completely wrapped by the monolayer and caused the monolayer to bulge, prominently, toward to the water phase, generating a large monolayer curvature. Thus, the interaction of C can inflict great disturbance to the monolayer structure.
Figure 1b indicates that C
6H
14O
2 and SiO
2 NPs still translocated across the pulmonary surfactant monolayer throughout the inhalation breathing process as they did throughout the exhalation breathing process. However, unlike in the exhalation breathing state, CaSO
4 penetrated the monolayer, although C NP still could not cross and was embedded into the monolayer.
The MSDs of the NPs and the centroid-to-centroid distances between the NPs and the monolayer during compression and expansion were calculated, respectively, as shown in
Figure 1c–f. In the exhalation breathing state, the MSD of SiO
2 was larger than those of the other three NPs, and the MSD of C was higher than that of CaSO
4 NP, indicating that the diffusion rate of the SiO
2 NP was the fastest, and C moved a longer distance than CaSO
4.
Figure 1e also shows that C and CaSO
4 NPs reached a relatively equilibrated state within approximately 10 ns, whereas C
6H
14O
2 and SiO
2 NPs took a longer time (i.e., around 45 ns). The initial centroid-to-centroid distance between the NP and the monolayer at time
t = 0 was set to 3.2 nm. The distance of the centroid position between the CaSO
4 NP and the monolayer at the equilibrium state was about 1.52 nm, and C was 0.27 nm away from the monolayer, meaning that C was embedded in the monolayer more deeply than CaSO
4. The final positions of C
6H
14O
2 and SiO
2 NPs were all approximately −3.3 nm away from the monolayer, with an average thickness of 2.08 nm, indicating that both NPs completely translocated across the monolayer. Compared with the compression process, the MSDs of all NPs during expansion were larger, which meant that all NPs could move faster, and their diffusion abilities were stronger than those in the exhalation breathing state. Another difference was that the MSD of CaSO
4 was larger than that of C, although SiO
2 showed the largest MSD.
Figure 1f shows that C
6H
14O
2 and SiO
2 NPs reached equilibrium within approximately 40 ns, which was shorter than that in the exhalation breathing state. Notably, the final position of the C NP was about 0.88 nm away from the centroid of the monolayer in the inhalation breathing state, indicating the C NP was embedded into the monolayer more deeply in the exhalation breathing state. These analysis results of MSD and the centroid-to-centroid distance confirmed our previous findings obtained from the molecular dynamics simulation snapshots.
The differences in hydrophilicity/hydrophobicity of the NPs explained the difference among the interaction behaviors. C NPs are typical hydrophobic NPs, and CaSO4 NP is semi-hydrophilic. C6H14O2 NPs and SiO2 NPs are strongly hydrophilic NPs, and SiO2 NPs are more hydrophilic.
The strong hydrophilicity of C6H14O2 and SiO2 NPs led to the strong attractive forces between the NPs and the hydrophilic head groups of DPPC molecules or between the NPs and the water molecules on the other side of the monolayer, which enabled the NPs to overcome the energy barrier and translocate across the monolayer. The uncharged and hydrophobic C NP was attracted by the hydrophobic tail groups of DPPC molecules, and its interaction with the hydrophilic head group was repulsive at the same time. Consequently, C cannot translocate across the monolayer, and its deep encapsulation inside the monolayer caused monolayer deformation.
For the semi-hydrophilic CaSO4 NP in the exhalation breathing state, the hydrophobic interaction between the NPs and hydrophobic apolar tail groups of DPPC molecules were not intensive. The interactions between the NP and hydrophilic head groups and water molecules were attractive but not sufficiently large for NP penetration, only causing the NP to stay in the surface region of the monolayer. In the inhalation breathing state, the reason for CaSO4 translocation across the monolayer may be that the relatively loose arrangement of lipid molecules enabled the attractive interaction between the NP and the monolayer to overcome the energy barrier, resulting in NP penetration through the expanded monolayer. We further elucidated this aspect by analyzing the surface area per lipid molecule and the energy component of the simulation system, as discussed in the following sections.
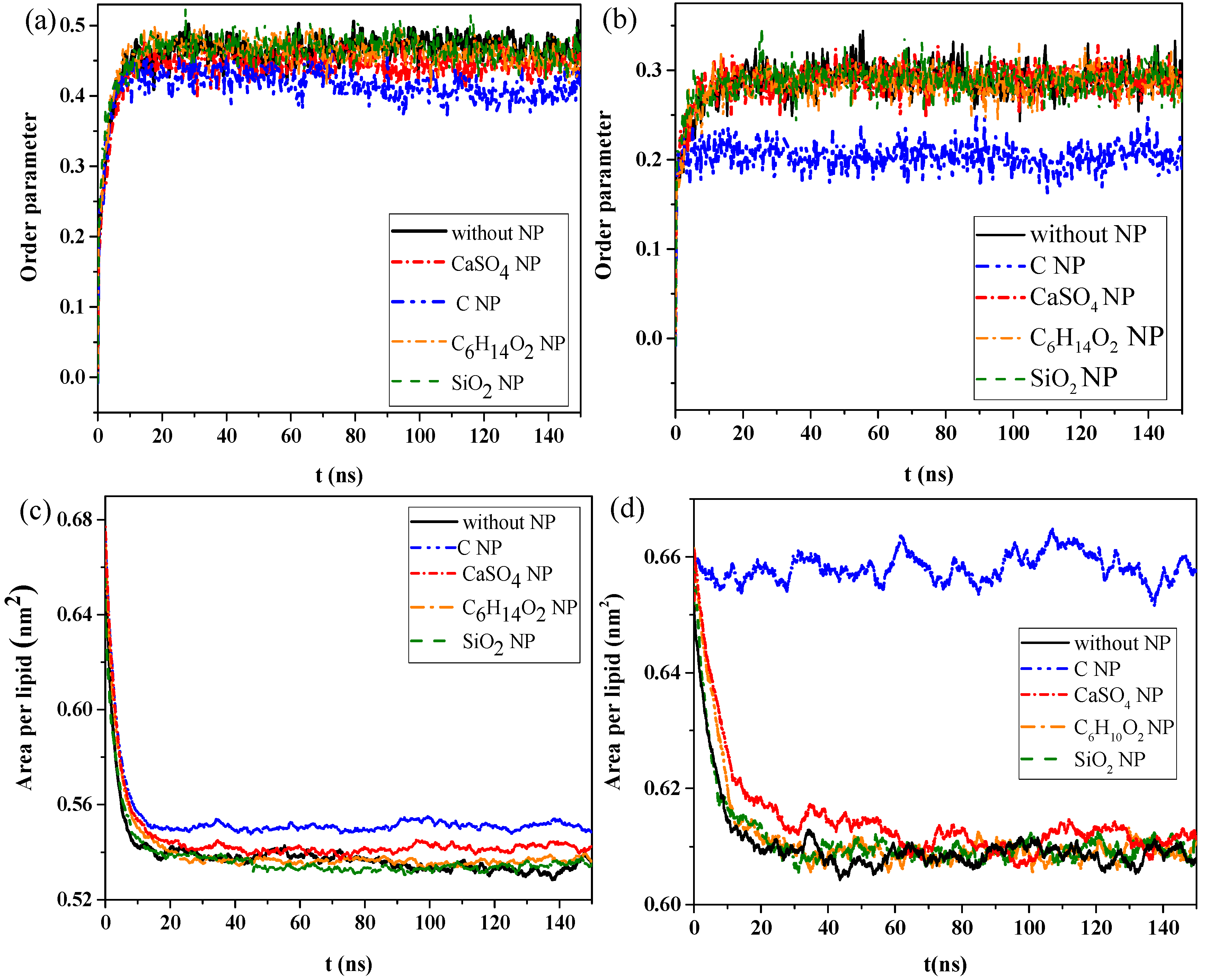
We then calculated the order parameters of lipid molecules in the pure monolayer system and NP monolayer systems over time, as well as the surface area per lipid molecule, to analyze the effect of interaction of NPs with the monolayer on the structural properties of the monolayer.
Figure 2a,b shows the changes in order parameters and surface area per lipid molecule in the exhalation and inhalation breathing states, respectively. A larger order parameter represented better consistency between the bonding and arrangement directions of the phospholipid monolayer, indicating better orderliness of the membrane lipid molecules.
Figure 2a shows that the order parameters of lipid molecules in the systems containing C
6H
14O
2 and SiO
2 were almost the same as those in the system without NPs, either in exhalation or at inhalation breathing states, all of which were approximately 0.458 or 0.285 (
Figure 2a,b) at equilibrium, respectively. Thus, C
6H
14O
2 and SiO
2 penetrations had a negligible effect on the orderliness of lipid molecules after NP membrane translocation. The order parameters for the system containing CaSO
4 NPs were 0.452 and 0.285 in exhalation or at inhalation breathing states, respectively, but they were 0.413 and 0.204 for C, indicating that the penetration of CaSO
4 had little effect on the monolayer structure in the inhalation breathing state. The presence of C most obviously affected the monolayer structure and led to decreased orderliness in the monolayer.
Figure 2c,d shows that the surface areas per lipid molecule in the systems without NPs and with C
6H
14O
2 and SiO
2 NPs were all about 0.53 nm
2 after equilibrium was reached. In other words, the effect of C
6H
14O
2 and SiO
2 on the monolayer structure was almost negligible. The surface areas per lipid molecule of the system containing C was about 0.552 and 0.657 nm
2 in exhalation or inhalation breathing states, respectively. These values were noticeably larger than those of other systems because the hydrophobicity of C NPs produced remarkable monolayer curvature and a large change in area. Analysis results of surface areas per lipid molecule were consistent with those of the order parameters. In general, the difference in interactions with the monolayer between SiO
2 and C
6H
14O
2 was not obvious because the differences in their hydrophilicity levels was not large.
2.2. Analyses of System Energy and Surface Pressure of the Monolayer
We calculated the total energy of different systems to compare the interactions of different NPs with the pulmonary surfactant monolayer in inhalation and exhalation breathing states and to further understand the mechanism underlying such interactions. All simulations in this study were performed in the NPT ensemble, and the total kinetic energy of the system remained unchanged considering the constant temperature under practical conditions. Thus, the change in total energy was equal to the change in potential energy.
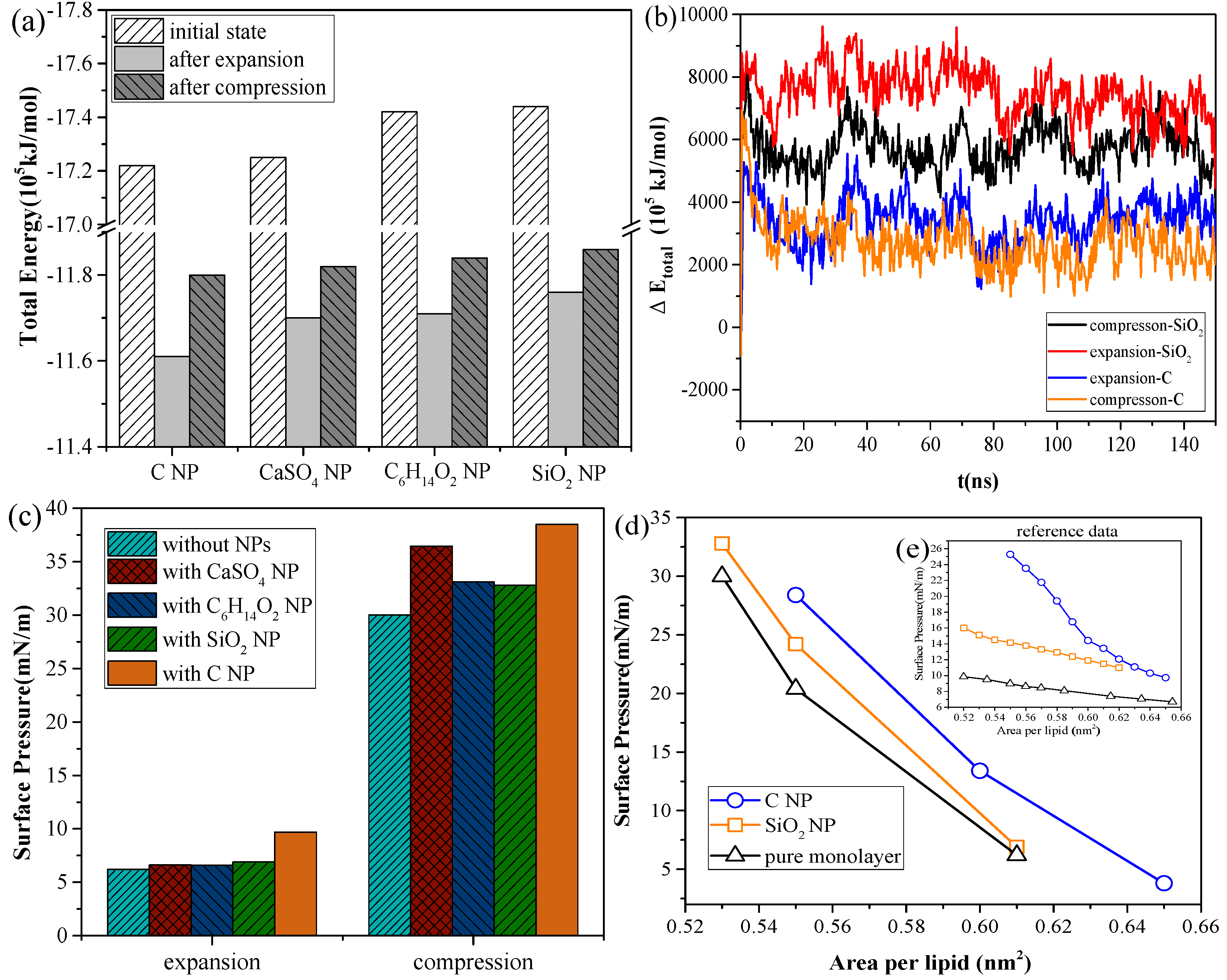
As shown in
Figure 3a, for the systems with the same NP, the total energy of the system in the inhalation breathing state was lower than that in the exhalation breathing state (i.e., the orderliness of the lipid molecules was more orderly, and the entropy of the system was lower). For all NPs, the total energies in both breathing states decreased after interacting with the monolayer, indicating that the interaction was irreversible. During the interaction process, the potential energy was converted to kinetic energy and was then dissipated by the simulation system to keep the total kinetic energy constant, so the potential energy of the system decreased. The NPs could not enter the air side again without applying external disturbance after it was embedded into the monolayer or crossed the membrane.
Figure 3b shows the difference in total energy between the NP monolayer system and the pure monolayer system, which includes the energy of the NP, the interaction energy between the NP and monolayer, and the interaction energy between the NP and water molecules. The change in this difference can reflect the change in NP potential energy, owing to the constant kinetic energy in the NPT ensemble. We also found SiO
2 had higher potential than C, indicating a larger interaction of SiO
2 with the lipid and water molecules and a greater likelihood of NP crossing the monolayer. The potential energy of SiO
2 in the inhalation breathing state was larger than that in the exhalation breathing state, indicating that the NP was subject to greater force and could more easily penetrate the monolayer.
The attractive interaction between the hydrophobic NP and hydrophobic groups of the monolayer was the dominant driving force acting on the NP during the process of NP movement and embedment, which had the same direction as the NP motion. Accordingly, the potential energy continually decreased until it eventually stabilized at a position where it was wrapped and a potential well formed. Conversely, hydrophilic NPs were subjected to a repulsive force, owing to the existence of a hydrophobic barrier produced by the lipid monolayer. The NP overcame the potential barrier and finally penetrated the monolayer when the hydrophilic interaction between the NP and water molecules was sufficiently strong. Otherwise, the NP was stopped by the potential barrier and embedded into the monolayer.
A stable, low surface tension of the pulmonary surfactant monolayer can maintain the stability of lung alveolars [
1,
20], but the surface tension may be affected by the interaction between the NPs and the monolayer. We calculated the surface pressure and pressure–area isotherm of the monolayer, which was commonly used to analyze the functional properties of the monolayer. The surface pressure π is given by π =
γw −
γ, where
γw and
γ represent the surface tension of pure water and the monolayer, respectively.
Figure 3c shows the changes in surface pressure of the monolayer in the exhalation and inhalation breathing states for the pure monolayer and NP monolayer systems after reaching the equilibrium state. Compared with the surface tension of the monolayer in the system without NPs, all interactions of different NPs with the monolayer resulted in decreased surface tension of the monolayer to a certain extent. The effects of all interactions in the exhalation breathing state were more remarkable than those in the inhalation breathing state, in which C showed the most pronounced effect on the surface tension of the monolayer. C was embedded into the monolayer because of its hydrophobic interaction with the hydrophobic tail chain of DPPC molecules and its interaction with the hydrophilic head groups of the lipid. This phenomenon resulted in decreased free area available for the lipid molecules and increased surface density of the monolayer. Thus, the surface tension of the monolayer was reduced and the surface pressure increased. For CaSO
4, although it was also embedded in the monolayer in the exhalation breathing state, the effect of the interaction on the surface pressure of the monolayer was still smaller than that of C because it had a smaller embedding degree, as shown in
Figure 1a. In the inhalation breathing state, the effect of the interaction of CaSO
4 with the monolayer was very slight, similar to those of C
6H
14O
2 and SiO
2 NPs, to their complete membrane translocation. These findings indicated that the breathing state played an important role in the interaction of NPs with the monolayer.
We compared the calculated surface pressure–area isothermal (π-A) of the monolayer for the strongly hydrophilic SiO
2 NP and the hydrophobic C NP with experimental data obtained from Ref. [
25] (
Figure 3d). Qualitative agreement was observed between the simulation and experimental results (i.e., strongly hydrophilic NPs had a smaller effect on the surface tension of the monolayer than the hydrophobic NPs). This finding was due to ability of the strongly hydrophilic NPs to translocate across the monolayer and not stay in it at equilibrium. The reason for the quantitative difference in simulation and experimental results was that the NPs initially mixed with DPPC for compression in the experiment, whereas the NPs were initially located on the air-side in the simulation and took more time to move to and interact with the monolayer.
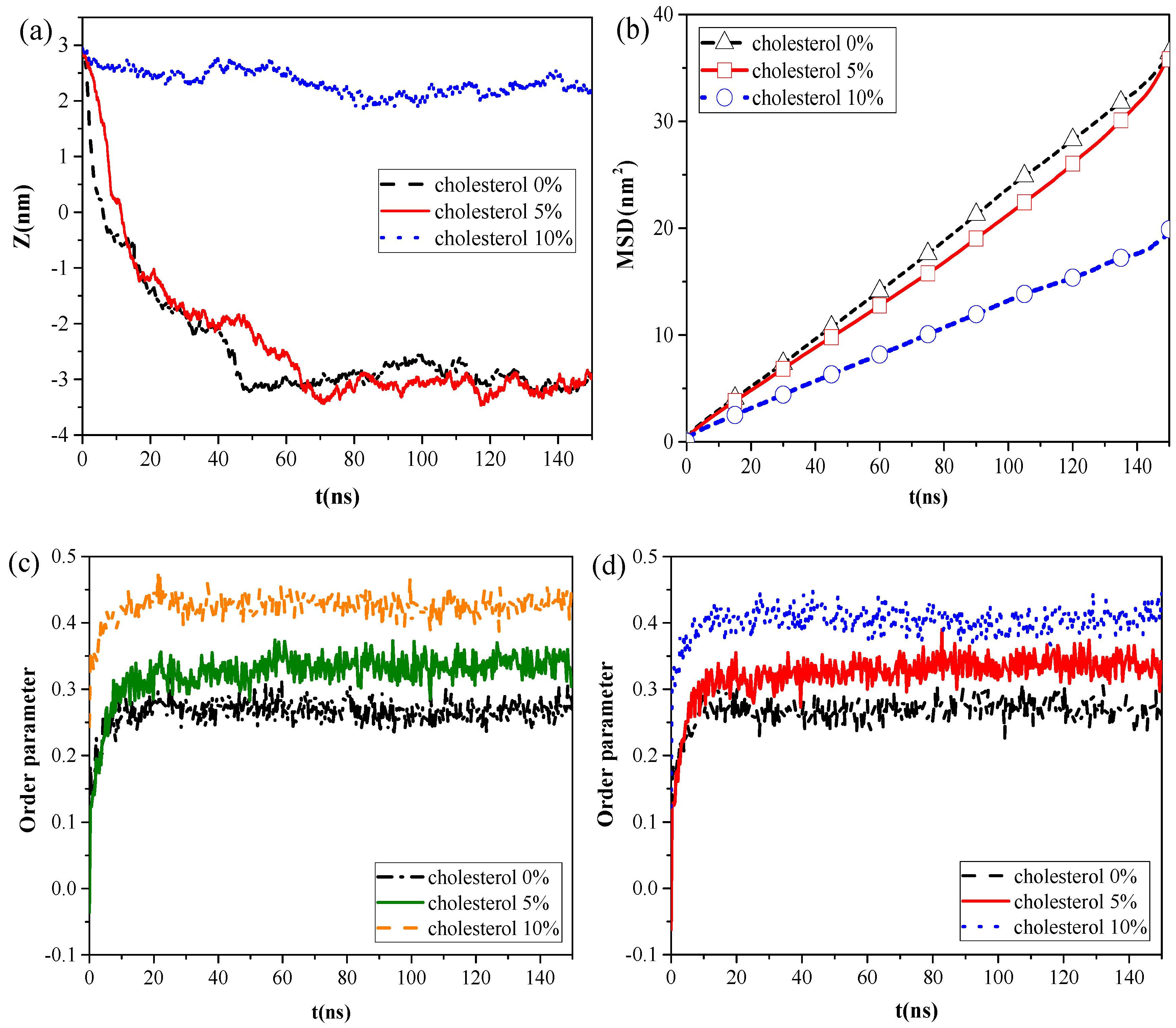
2.3. Effect of Cholestrol Content of the Monolayer and NP Structural Properties on the Interaction
Cholesterol is reportedly present in the lipid membrane, at 5%–10% by mass, and constitutes the major fraction of neutral lipids [
3]. To investigate the effect of cholesterol content on the interaction of NPs with the pulmonary surfactant monolayer, we established CG models of the pulmonary membrane with 0%, 5%, and 10% cholesterol content, respectively, and simulated its interaction with C, CaSO
4, or SiO
2 NPs. We found that all kinds of NPs moved slower with increased cholesterol content, and that C could not penetrate all three kinds of the monolayers at the inhalation breathing state, whereas SiO
2 could cross all these monolayers.
However, the calculation results of the centroid–centroid distance of the NP with the monolayer and the MSD of the lipid molecules (
Figure 4a,b) revealed that CaSO
4 can translocate only across the monolayer with 0% and 5% cholesterol content, and the MSD of the lipid molecules prominently decreased in the system with 10% cholesterol content. We then analyzed the order parameters of the lipid molecules of the monolayer with 0%, 5%, and 10% cholesterol contents, respectively, in the systems without NPs and with CaSO
4 (
Figure 4c,d). An increase in cholesterol content of the monolayer led to an increase in the order parameters of the monolayer. The fluidity of lipid molecules is usually related to their structural ordering and is manifested in the tail of phospholipid molecules [
26]. Our results indicated that the cholesterol content remarkably influenced the fluidity of the pulmonary surfactant monolayer and that increased cholesterol content led to a more orderly arrangement of lipid molecules. This ordering enhanced the packing of the hydrophobic portion of the lipid molecule and, thus, reduced the fluidity of the pulmonary surfactant monolayer. Subsequently, the NPs encountered increased difficulty in penetrating the monolayer. CaSO
4 could cross the monolayers with 0% and 5% cholesterol content, so little difference in order parameter between the pure monolayer and CaSO
4 NP monolayer systems was observed. For the lipid molecules of the monolayer with 10% cholesterol content, the order parameters of the two systems were 0.40 and 0.42, respectively. This finding indicated that the difference was not very remarkable because CaSO
4 stayed only on the surface region of the monolayer, and it inflicted slight disturbance to the monolayer structure because the centroid–centroid distance between the NP with 3 nm diameter and the monolayer with 2 nm thickness was only 2.23 nm.
We analyzed the effects of NP structural properties on the interaction between NPs and the pulmonary surfactant monolayer, taking the effects of size and shape into account.
Figure 5a shows the centroid-to-centroid distances between a hydrophilic and spherical NP without charge and surfactant monolayer and the corresponding snapshots at equilibrium under different NP size conditions, respectively. Smaller NPs took shorter times to cross the monolayer and reach the equilibrium state, indicating that the difficulty of NP membrane translocation was positively correlated with NP size. Notably, all NPs attached onto the hydrophilic side of the monolayer after membrane translocation, and no detachment from the monolayer occurred due to the attractive interaction of the hydrophilic head groups of the monolayer with the NPs. Although all hydrophilic NPs can successfully cross the monolayer, the NPs did not stay in a certain position and remain fixed, and the smaller NPs were more easily affected by thermal fluctuations and had a larger fluctuation range.
Three types of neutral hydrophilic NPs with three common shapes for NPs in the air, namely, spherical (diameter d = 3 nm), rod-shaped (diameter d = 3 nm and height h = 3 nm), and flaky (size d = 3 nm and thickness δ = 0.5 nm), were modeled to analyze the effect of NP shape on the interaction. As shown in
Figure 5b, all three types of NPs can translocate across the monolayer. The difference was that the spherical and rod-shaped NPs still adsorbed onto the monolayer at equilibrium, whereas the flaky NP penetrated through and completely separated from the monolayer, entering into the water phase. During NP translocation, we observed that the angles between the monolayer and rod-shaped or flaky NPs were continuously adjusted according to the changes in their relative position and the variation in their interactions. The rod-shaped NP contacted and penetrated the monolayer with the long axis perpendicular to the plane of the monolayer from the initial parallel placement. Similarly, the flaky NP spontaneously moved and rotated from the initial placement in parallel with the monolayer to the vertical placement with a minimum contact area made with the monolayer. After the NP was embedded into the monolayer, the NP eventually detached from the monolayer because attachment of the NP to hydrophilic head groups of the monolayer cannot counteract the attractive force between the NP and water molecules due to minimal contact.
To quantify the difference in penetration behaviors of NPs with different shapes, we calculated the centroid-to-centroid distance between NPs and the monolayer, and we found that rod-shaped NPs took less time (t = 17 ns) to cross the monolayer and reach the equilibrium state than spherical NPs (t = 21 ns), although all of them eventually attached onto the monolayer and could not detach from it. The flaky NP spent more time (t = 50 ns) to penetrate the pulmonary surfactant monolayer, and equilibrium was reached within about 90 ns, where it then completely entered the water phase and was surrounded by water molecules.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





