The Biological Axis of Protein Arginine Methylation and Asymmetric Dimethylarginine



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. PRMTs
2.1. PRMT Family in Mammalian Cells
2.2. Novel Arginine Methyltransferases
3. Regulatory Effects of Arginine Methylation on Protein Recognition and Function
3.1. Protein-Nucleic Interactions
3.2. Protein-Protein Interactions
3.3. Liquid-Liquid Phase Separation
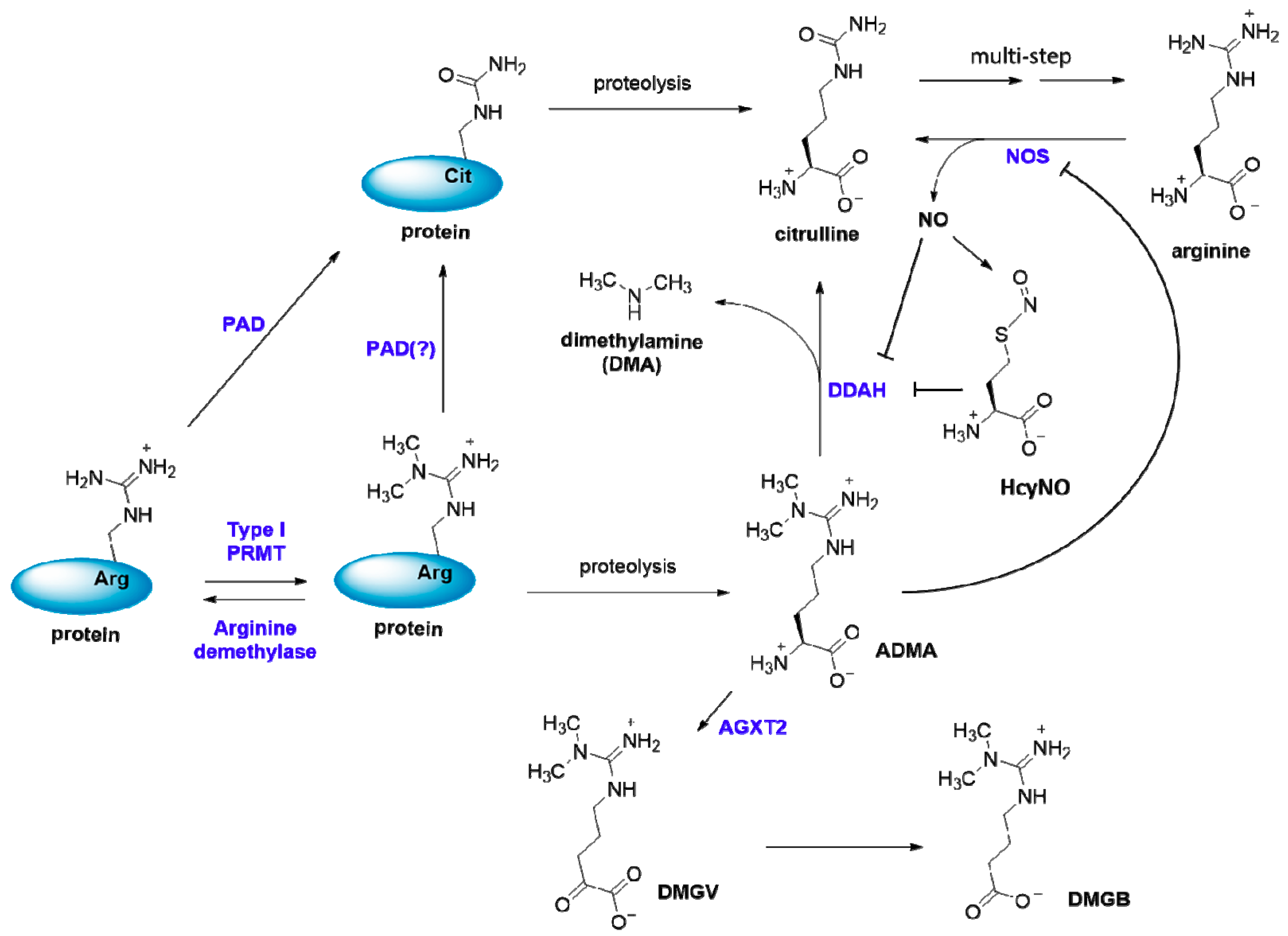
4. ADMA—the Small Molecule Metabolite Product of Arginine Methylation
4.1. ADMA Production
4.2. ADMA Metabolism and Excretion
4.3. Inhibition of NOS by ADMA
4.4. Physiological Impact of ADMA
4.5. Medicinal Significance of PRMT Inhibitors in ADMA-caused Cardiovascular Disorders
5. Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 53BP1 | TP53-binding protein 1 |
| ADMA | The small molecule metabolite NG, NG-dimethylarginine (asymmetric dimethylarginine) |
| AGXT2 | Alanine-glyoxylate aminotransferase 2 |
| BRCA1 | Breast cancer type 1 susceptibility protein |
| BRCT | BRCA1 C-terminal domain |
| CA150 | Transcription factor CA150 |
| CARM1 | Coactivator associated arginine methyltransferase 1 |
| CCR2 | C-C chemokine receptor type 2 |
| CVD | Cardiovascular disease |
| CXCR2 | C-X-C chemokine receptor type 2 |
| DDAH | Dimethylarginine dimethylaminohydrolase |
| Ddx4 | DEAD box protein 4 |
| DMGB | γ-(NG,NG-dimethylguanidino)butyric acid |
| DMGV | α-keto-δ-(NG,NG-dimethylguanidino)valeric acid |
| ERα | Estrogen receptor alpha |
| ERE | Estrogen response element |
| EWS | Ewing’s sarcoma protein |
| FOXO1 | Forkhead box protein O1 |
| FUS | Fused in sarcoma |
| G3BP1 | Ras-GTPase activating SH3 domain-binding protein 1 |
| HcyNO | S-nitroso-l-homocysteine |
| HSP70 | Heat-shock protein of 70 kDa |
| IL-8 | Interleukin-8 |
| JMJD | JmjC-domain-containing protein |
| LLPS | Liquid-liquid phase separation |
| l-NMMA | The small molecule NG-monomethyl-l-arginine (MMA) |
| Lsm | Sm-like protein |
| MCP-1 | Monocyte chemoattractant protein-1 |
| Mettl23 | Methyltransferase-like protein 23 |
| MidA | Mitochondrial dysfunction protein A |
| MLL | Mixed lineage leukemia protein |
| NDUFAF7 | NADH: ubiquinone oxidoreductase complex assembly factor 7 |
| NDUFS2 | NADH: ubiquinone oxidoreductase core subunit S2 |
| NF-κB | Nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| Pax7 | Paired box protein 7 |
| P-bodies | Processing bodies |
| p300 | Histone acetyltransferase p300 |
| PRMT | Protein N-arginine methyltransferase |
| PRMTi | PRMT inhibitor |
| PTM | Post-translational modification |
| RBP | RNA-binding protein |
| RHA | RNA helicase A |
| Rme1 | Monomethylarginine residue in proteins |
| Rme2a | Asymmetric dimethylarginine residue in proteins |
| Rme2s | Symmetric dimethylarginine residue in proteins |
| SAH | S-adenosyl-l-homocysteine |
| SAM | S-adenosyl-l-methionine (AdoMet) |
| SDMA | The small molecule NG, N’G-dimethylarginine (symmetric dimethylarginine) |
| Sm | “Smith,” small nuclear ribonucleoprotein-associated protein |
| SMN | Survival of motor neurons protein |
| snRNP | Small nuclear ribonucleoprotein |
| SPF30 | Survival of motor neuron-related-splicing factor 30 |
| TAR | trans-activation response element |
| TDRD | Tudor domain-containing protein |
| TIA-1 | T-cell intracellular antigen-1 |
| TIA-R | TIA-1 related protein |
| TNF-α | Tumor necrosis factor alpha |
| TML | 6-N-trimethyllysine |
| TRAF 6 | TNF receptor-associated factor 6 |
| UUO | Unilateral ureter obstruction |
References
- Migliori, V.; Phalke, S.; Bezzi, M.; Guccione, E. Arginine/lysine-methyl/methyl switches: Biochemical role of histone arginine methylation in transcriptional regulation. Epigenomics 2010, 2, 119–137. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci. Signal. 2016, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical Biology of Protein Arginine Modifications in Epigenetic Regulation. Chem. Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulau, P.; Zakrzewicz, D.; Kitowska, K.; Wardega, B.; Kreuder, J.; Eickelberg, O. Quantitative assessment of arginine methylation in free versus protein-incorporated amino acids in vitro and in vivo using protein hydrolysis and high-performance liquid chromatography. BioTechniques 2006, 40, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Uhlmann, T.; Geoghegan, V.L.; Thomas, B.; Ridlova, G.; Trudgian, D.C.; Acuto, O. A method for large-scale identification of protein arginine methylation. Mol. Cell. Proteomics 2012, 11, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Morales, Y.; Cáceres, T.; May, K.; Hevel, J.M. Biochemistry and regulation of the protein arginine methyltransferases (PRMTs). Arch. Biochem. Biophys. 2016, 590, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Fulton, M.D.; Brown, T.; Zheng, Y.G. Mechanisms and Inhibitors of Histone Arginine Methylation. Chem. Rec. 2018, 18, 1792–1807. [Google Scholar] [CrossRef]
- Dilworth, D.; Barsyte-Lovejoy, D. Targeting protein methylation: From chemical tools to precision medicines. Cell. Mol. Life Sci. 2019, 1–9. [Google Scholar] [CrossRef]
- Guo, A.; Gu, H.; Zhou, J.; Mulhern, D.; Wang, Y.; Lee, K.A.; Yang, V.; Aguiar, M.; Kornhauser, J.; Jia, X.; et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 2014, 13, 372–387. [Google Scholar] [CrossRef]
- Lott, K.; Li, J.; Fisk, J.C.; Wang, H.; Aletta, J.M.; Qu, J.; Read, L.K. Global proteomic analysis in trypanosomes reveals unique proteins and conserved cellular processes impacted by arginine methylation. J. Proteomics 2013, 91, 210–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Wong, C.C. The story of protein arginine methylation: Characterization, regulation, and function. Expert Rev. Proteomics 2017, 14, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, M.R.; Scherer, C.A.; Chen, J.; Roshon, M.J.; Ruley, H.E. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell. Biol. 2000, 20, 4859–4869. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Vemulapalli, V.; Patananan, A.N.; Huang, G.L.; Di Lorenzo, A.; Richard, S.; Comb, M.J.; Guo, A.; Clarke, S.G.; Bedford, M.T. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci. Rep. 2013, 3, 1311. [Google Scholar] [CrossRef] [PubMed]
- Hadjikyriacou, A.; Yang, Y.; Espejo, A.; Bedford, M.T.; Clarke, S.G. Unique Features of Human Protein Arginine Methyltransferase 9 (PRMT9) and Its Substrate RNA Splicing Factor SF3B2. J. Biol. Chem. 2015, 290, 16723–16743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrossian, T.C.; Clarke, S.G. Uncovering the human methyltransferasome. Mol. Cell. Proteomics 2011, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef]
- Hadjikyriacou, A.; Clarke, S.G. Caenorhabditis elegans PRMT-7 and PRMT-9 Are Evolutionarily Conserved Protein Arginine Methyltransferases with Distinct Substrate Specificities. Biochemistry 2017, 56, 2612–2626. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Fuhrmann, J.; Thompson, P.R. Protein Arginine Methyltransferase 5 Catalyzes Substrate Dimethylation in a Distributive Fashion. Biochemistry 2014, 53, 7884–7892. [Google Scholar] [CrossRef]
- Yang, Y.; Hadjikyriacou, A.; Xia, Z.; Gayatri, S.; Kim, D.; Zurita-Lopez, C.; Kelly, R.; Guo, A.; Li, W.; Clarke, S.G.; et al. PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat. Commun. 2015, 6, 6428. [Google Scholar] [CrossRef] [PubMed]
- Zurita-Lopez, C.I.; Sandberg, T.; Kelly, R.; Clarke, S.G. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming omega-NG-monomethylated arginine residues. J. Biol. Chem. 2012, 287, 7859–7870. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Hadjikyriacou, A.; Clarke, S.G. Substrate specificity of human protein arginine methyltransferase 7 (PRMT7): The importance of acidic residues in the double E loop. J. Biol. Chem. 2014, 289, 32604–32616. [Google Scholar] [CrossRef] [PubMed]
- Zurita Rendon, O.; Silva Neiva, L.; Sasarman, F.; Shoubridge, E.A. The arginine methyltransferase NDUFAF7 is essential for complex I assembly and early vertebrate embryogenesis. Hum. Mol. Genet. 2014, 23, 5159–5170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhein, V.F.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. NDUFAF7 methylates arginine 85 in the NDUFS2 subunit of human complex I. J. Biol. Chem. 2013, 288, 33016–33026. [Google Scholar] [CrossRef]
- Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Post-translational modifications near the quinone binding site of mammalian complex I. J. Biol. Chem. 2013, 288, 24799–24808. [Google Scholar] [CrossRef]
- Hameed, U.F.S.; Sanislav, O.; Lay, S.T.; Annesley, S.J.; Jobichen, C.; Fisher, P.R.; Swaminathan, K.; Arold, S.T. Proteobacterial Origin of Protein Arginine Methylation and Regulation of Complex I Assembly by MidA. Cell Rep. 2018, 24, 1996–2004. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, Y.; Tsusaka, T.; Shimizu, N.; Morita, K.; Suzuki, T.; Machida, S.; Satoh, M.; Honda, A.; Hirose, M.; Kamimura, S.; et al. Histone H3 Methylated at Arginine 17 Is Essential for Reprogramming the Paternal Genome in Zygotes. Cell Rep. 2017, 20, 2756–2765. [Google Scholar] [CrossRef] [Green Version]
- Chong, P.A.; Vernon, R.M.; Forman-Kay, J.D. RGG/RG Motif Regions in RNA Binding and Phase Separation. J. Mol. Biol. 2018, 430, 4650–4665. [Google Scholar] [CrossRef]
- Lorton, B.M.; Shechter, D. Cellular consequences of arginine methylation. Cell. Mol. Life Sci. 2019, 1–24. [Google Scholar] [CrossRef]
- Liu, Q.; Dreyfuss, G. In vivo and in vitro arginine methylation of RNA-binding proteins. Mol. Cell. Biol. 1995, 15, 2800–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajpurohit, R.; Paik, W.K.; Kim, S. Effect of enzymic methylation of heterogeneous ribonucleoprotein particle A1 on its nucleic-acid binding and controlled proteolysis. Biochem. J. 1994, 304, 903–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahama, K.; Kino, K.; Arai, S.; Kurokawa, R.; Oyoshi, T. Identification of Ewing’s sarcoma protein as a G-quadruplex DNA- and RNA-binding protein. FEBS J. 2011, 278, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Chiu, W.C.; Yao, Y.C.; Cheng, R.P. Effect of arginine methylation on the RNA recognition and cellular uptake of Tat-derived peptides. Bioorg. Med. Chem. 2015, 23, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Friesen, W.J.; Massenet, S.; Paushkin, S.; Wyce, A.; Dreyfuss, G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001, 7, 1111–1117. [Google Scholar] [CrossRef]
- Brahms, H.; Meheus, L.; de Brabandere, V.; Fischer, U.; Luhrmann, R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B’ and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA (New York, NY) 2001, 7, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Cote, J.; Shaaban, S.; Bedford, M.T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell 2007, 25, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Lu, Y.; Espejo, A.; Wu, J.; Xu, W.; Liang, S.; Bedford, M.T. TDRD3 is an effector molecule for arginine-methylated histone marks. Mol. Cell 2010, 40, 1016–1023. [Google Scholar] [CrossRef]
- Liu, K.; Guo, Y.; Liu, H.; Bian, C.; Lam, R.; Liu, Y.; Mackenzie, F.; Rojas, L.A.; Reinberg, D.; Bedford, M.T.; et al. Crystal structure of TDRD3 and methyl-arginine binding characterization of TDRD3, SMN and SPF30. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef]
- Goulet, I.; Boisvenue, S.; Mokas, S.; Mazroui, R.; Cote, J. TDRD3, a novel Tudor domain-containing protein, localizes to cytoplasmic stress granules. Hum. Mol. Genet. 2008, 17, 3055–3074. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.H.; Bedford, M.T.; Stallcup, M.R. Regulated recruitment of tumor suppressor BRCA1 to the p21 gene by coactivator methylation. Genes Dev. 2011, 25, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabe, Y.I.; Wang, Y.X.; McKinnell, I.W.; Bedford, M.T.; Rudnicki, M.A. Carm1 Regulates Pax7 Transcriptional Activity through MLL1/2 Recruitment during Asymmetric Satellite Stem Cell Divisions. Cell Stem Cell 2012, 11, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayatri, S.; Bedford, M.T. Readers of histone methylarginine marks. Biochim. Biophys. Acta 2014, 1839, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdile, V.; De Paola, E.; Paronetto, M.P. Aberrant Phase Transitions: Side Effects and Novel Therapeutic Strategies in Human Disease. Front. Genet. 2019, 10, 173. [Google Scholar] [CrossRef] [PubMed]
- Hofweber, M.; Dormann, D. Friend or foe-Post-translational modifications as regulators of phase separation and RNP granule dynamics. J. Biol. Chem. 2019, 294, 7137–7150. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef] [PubMed]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef]
- Qamar, S.; Wang, G.Z.; Randle, S.J.; Ruggeri, F.S.; Varela, J.A.; Lin, J.Q.; Phillips, E.C.; Miyashita, A.; Williams, D.; Strohl, F.; et al. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell 2018, 173, 720–734. [Google Scholar] [CrossRef]
- Chen, Y.H.; Xu, X.; Sheng, M.J.; Zhang, X.Y.; Gu, Q.; Zheng, Z. PRMT-1 and DDAHs-induced ADMA upregulation is involved in ROS- and RAS-mediated diabetic retinopathy. Exp. Eye Res. 2009, 89, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Yokoro, M.; Suzuki, M.; Murota, K.; Otsuka, C.; Yamashita, H.; Takahashi, Y.; Tsuji, H.; Kimoto, M. Asymmetric dimethylarginine, an endogenous NOS inhibitor, is actively metabolized in rat erythrocytes. Biosci. Biotechnol. Biochem. 2012, 76, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.M.; Vallance, P. The synthesis and metabolism of asymmetric dimethylarginine (ADMA). Eur. J. Clin. Pharmacol. 2006, 62, 33–38. [Google Scholar] [CrossRef]
- Vallance, P.; Leiper, J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1023–1030. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Lin, P.; Li, L.; Chen, D.; Yang, X.; Xu, L.; Zhou, B.; Wang, C.; Zhang, Y.; Luo, C.; et al. Reduced asymmetric dimethylarginine accumulation through inhibition of the type I protein arginine methyltransferases promotes renal fibrosis in obstructed kidneys. FASEB J. 2019, 33, 6948–6956. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.B.; Sui, G.G.; Lu, X.Y.; Sun, Z.L. Elevated Levels of ADMA Are Associated with Lower DDAH2 and Higher PRMT1 in LPS-Induced Endometritis Rats. Inflammation 2018, 41, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, Y.; Yang, T.; Wang, Y.; Bai, Y.; Xie, X. ADMA induces monocyte adhesion via activation of chemokine receptors in cultured THP-1 cells. Cytokine 2008, 43, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Niu, M.; Yin, S.; Zhang, F.; Shi, R. Nephroprotective effects of nebivolol in 2K1C rats through regulation of the kidney ROS-ADMA-NO pathway. Pharmacol. Rep. 2018, 70, 917–929. [Google Scholar] [CrossRef]
- Jiang, J.L.; Zhang, X.H.; Li, N.S.; Rang, W.Q.; Feng, Y.; Hu, C.P.; Li, Y.J.; Deng, H.W. Probucol decreases asymmetrical dimethylarginine level by alternation of protein arginine methyltransferase I and dimethylarginine dimethylaminohydrolase activity. Cardiovasc. Drugs Ther. 2006, 20, 281–294. [Google Scholar] [CrossRef]
- Servillo, L.; Giovane, A.; Cautela, D.; Castaldo, D.; Balestrieri, M.L. The methylarginines NMMA, ADMA, and SDMA are ubiquitous constituents of the main vegetables of human nutrition. Nitric Oxide 2013, 30, 43–48. [Google Scholar] [CrossRef]
- Nijveldt, R.J.; Teerlink, T.; van Guldener, C.; Prins, H.A.; van Lambalgen, A.A.; Stehouwer, C.D.; Rauwerda, J.A.; van Leeuwen, P.A. Handling of asymmetrical dimethylarginine and symmetrical dimethylarginine by the rat kidney under basal conditions and during endotoxaemia. Nephrol. Dial. Transplant. 2003, 18, 2542–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijveldt, R.J.; Teerlink, T.; Siroen, M.P.C.; Van Lambalgen, A.A.; Rauwerda, J.A.; Van Leuuwen, P.A.M. The liver is an important organ in the metabolism of asymmetrical dimethylarginine (ADMA). Clin. Nutr. 2003, 22, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gill, P.S.; Chabrashvili, T.; Onozato, M.L.; Raggio, J.; Mendonca, M.; Dennehy, K.; Li, M.; Modlinger, P.; Leiper, J.; et al. Isoform-specific regulation by N(G),N(G)-dimethylarginine dimethylaminohydrolase of rat serum asymmetric dimethylarginine and vascular endothelium-derived relaxing factor/NO. Circ. Res. 2007, 101, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Purification and properties of a new enzyme, NG,NG-dimethylarginine dimethylaminohydrolase, from rat kidney. J. Biol. Chem. 1989, 264, 10205–10209. [Google Scholar] [PubMed]
- Murray-Rust, J.; Leiper, J.; McAlister, M.; Phelan, J.; Tilley, S.; Maria, J.S.; Vallance, P.; McDonald, N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat. Struct. Biol. 2001, 8, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.; Murray-Rust, J.; McDonald, N.; Vallance, P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: Further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc. Natl. Acad. Sci. USA 2002, 99, 13527–13532. [Google Scholar] [CrossRef] [PubMed]
- Hong, L.; Fast, W. Inhibition of human dimethylarginine dimethylaminohydrolase-1 by S-nitroso-L-homocysteine and hydrogen peroxide. Analysis, quantification, and implications for hyperhomocysteinemia. J. Biol. Chem. 2007, 282, 34684–34692. [Google Scholar] [CrossRef] [PubMed]
- Knipp, M.; Braun, O.; Vasak, M. Searching for DDAH inhibitors: S-nitroso-L-homocysteine is a chemical lead. J. Am. Chem. Soc. 2005, 127, 2372–2373. [Google Scholar] [CrossRef]
- Ogawa, T.; Kimoto, M.; Watanabe, H.; Sasaoka, K. Metabolism of Ng,Ng-Dimethylarginine and Ng,N’g-Dimethylarginine in Rats. Arch. Biochem. Biophys. 1987, 252, 526–537. [Google Scholar] [CrossRef]
- Rodionov, R.N.; Murry, D.J.; Vaulman, S.F.; Stevens, J.W.; Lentz, S.R. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J. Biol. Chem. 2010, 285, 5385–5391. [Google Scholar] [CrossRef]
- Caplin, B.; Wang, Z.; Slaviero, A.; Tomlinson, J.; Dowsett, L.; Delahaye, M.; Salama, A.; International Consortium for Blood Pressure Genome-Wide Association, Studies; Wheeler, D.C.; Leiper, J. Alanine-glyoxylate aminotransferase-2 metabolizes endogenous methylarginines, regulates NO, and controls blood pressure. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2892–2900. [Google Scholar] [CrossRef] [PubMed]
- Rodionov, R.N.; Jarzebska, N.; Weiss, N.; Lentz, S.R. AGXT2: A promiscuous aminotransferase. Trends Pharmacol. Sci. 2014, 35, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone deimination antagonizes arginine methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Kearney, P.L.; Bhatia, M.; Jones, N.G.; Yuan, L.; Glascock, M.C.; Catchings, K.L.; Yamada, M.; Thompson, P.R. Kinetic characterization of protein arginine deiminase 4: A transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005, 44, 10570–10582. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.R.; Fast, W. Histone citrullination by protein arginine deiminase: Is arginine methylation a green light or a roadblock? ACS Chem. Biol. 2006, 1, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Tikhanovich, I.; Kuravi, S.; Artigues, A.; Villar, M.T.; Dorko, K.; Nawabi, A.; Roberts, B.; Weinman, S.A. Dynamic Arginine Methylation of Tumor Necrosis Factor (TNF) Receptor-associated Factor 6 Regulates Toll-like Receptor Signaling. J. Biol. Chem. 2015, 290, 22236–22249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.C.; Reineke, L.C.; Jain, A.; Jung, S.Y.; Lloyd, R.E. Histone arginine demethylase JMJD6 is linked to stress granule assembly through demethylation of the stress granule-nucleating protein G3BP1. J. Biol. Chem. 2017, 292, 18886–18896. [Google Scholar] [CrossRef]
- Gao, W.W.; Xiao, R.Q.; Peng, B.L.; Xu, H.T.; Shen, H.F.; Huang, M.F.; Shi, T.T.; Yi, J.; Zhang, W.J.; Wu, X.N.; et al. Arginine methylation of HSP70 regulates retinoid acid-mediated RARbeta2 gene activation. Proc. Natl. Acad. Sci. USA 2015, 112, 3327–3336. [Google Scholar] [CrossRef]
- Lawrence, P.; Conderino, J.S.; Rieder, E. Redistribution of demethylated RNA helicase A during foot-and-mouth disease virus infection: Role of Jumonji C-domain containing protein 6 in RHA demethylation. Virology 2014, 452, 1–11. [Google Scholar] [CrossRef]
- Poulard, C.; Rambaud, J.; Hussein, N.; Corbo, L.; Le Romancer, M. JMJD6 regulates ERalpha methylation on arginine. PLoS ONE 2014, 9, e87982. [Google Scholar] [CrossRef] [PubMed]
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R., 3rd; et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009, 325, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Li, J.J.; Wang, Y.Q.; Li, X.; Mao, H.L.; Liu, Y.F.; Chen, C.D. The hydroxylation activity of Jmjd6 is required for its homo-oligomerization. J. Cell. Biochem. 2012, 113, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Mantri, M.; Loik, N.D.; Hamed, R.B.; Claridge, T.D.; McCullagh, J.S.; Schofield, C.J. The 2-oxoglutarate-dependent oxygenase JMJD6 catalyses oxidation of lysine residues to give 5S-hydroxylysine residues. Chembiochem 2011, 12, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Bottger, A.; Islam, M.S.; Chowdhury, R.; Schofield, C.J.; Wolf, A. The oxygenase Jmjd6—A case study in conflicting assignments. Biochem. J. 2015, 468, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.; Wegener, I.; Burrells, A.; Bose, J.; Wolf, A.; Erck, C.; Butler, D.; Schofield, C.J.; Bottger, A.; Lengeling, A. Analysis of Jmjd6 cellular localization and testing for its involvement in histone demethylation. PLoS ONE 2010, 5, e13769. [Google Scholar] [CrossRef]
- Li, S.; Ali, S.; Duan, X.; Liu, S.; Du, J.; Liu, C.; Dai, H.; Zhou, M.; Zhou, L.; Yang, L.; et al. JMJD1B Demethylates H4R3me2s and H3K9me2 to Facilitate Gene Expression for Development of Hematopoietic Stem and Progenitor Cells. Cell Rep. 2018, 23, 389–403. [Google Scholar] [CrossRef] [Green Version]
- Walport, L.J.; Hopkinson, R.J.; Chowdhury, R.; Schiller, R.; Ge, W.; Kawamura, A.; Schofield, C.J. Arginine demethylation is catalysed by a subset of JmjC histone lysine demethylases. Nat. Commun. 2016, 7, 11974. [Google Scholar] [CrossRef]
- Zhang, J.; Jing, L.; Li, M.; He, L.; Guo, Z. Regulation of histone arginine methylation/demethylation by methylase and demethylase (Review). Mol. Med. Rep. 2019, 19, 3963–3971. [Google Scholar] [CrossRef]
- Wesche, J.; Kuhn, S.; Kessler, B.M.; Salton, M.; Wolf, A. Protein arginine methylation: A prominent modification and its demethylation. Cell. Mol. Life Sci. 2017, 74, 3305–3315. [Google Scholar] [CrossRef]
- Zakrzewicz, D.; Eickelberg, O. From arginine methylation to ADMA: A novel mechanism with therapeutic potential in chronic lung diseases. BMC Pulm. Med. 2009, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Cardounel, A.J.; Cui, H.; Samouilov, A.; Johnson, W.; Kearns, P.; Tsai, A.L.; Berka, V.; Zweier, J.L. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J. Biol. Chem. 2007, 282, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.; Nandi, M.; Torondel, B.; Murray-Rust, J.; Malaki, M.; O’Hara, B.; Rossiter, S.; Anthony, S.; Madhani, M.; Selwood, D.; et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med. 2007, 13, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Dayoub, H.; Achan, V.; Adimoolam, S.; Jacobi, J.; Stuehlinger, M.C.; Wang, B.Y.; Tsao, P.S.; Kimoto, M.; Vallance, P.; Patterson, A.J.; et al. Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: Genetic and physiological evidence. Circulation 2003, 108, 3042–3047. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [PubMed]
- Betz, B.; Moller-Ehrlich, K.; Kress, T.; Kniepert, J.; Schwedhelm, E.; Boger, R.H.; Wanner, C.; Sauvant, C.; Schneider, R. Increased symmetrical dimethylarginine in ischemic acute kidney injury as a causative factor of renal L-arginine deficiency. Transl. Res. 2013, 162, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Telo, S.; Kirkil, G.; Kuluozturk, M.; Balin, M.; Deveci, F. Can ADMA play a role in determining pulmonary hypertension related to chronic obstructive pulmonary disease? Clin. Respir. J. 2018, 12, 1433–1438. [Google Scholar] [CrossRef]
- Di Franco, M.; Lucchino, B.; Conti, F.; Valesini, G.; Spinelli, F.R. Asymmetric Dimethyl Arginine as a Biomarker of Atherosclerosis in Rheumatoid Arthritis. Mediators Inflamm. 2018, 2018, 3897295. [Google Scholar] [CrossRef]
- Boger, R.H. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J. Nutr. 2004, 134, 2842–2847. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Böger, R.H. The role of asymmetric and symmetric dimethylarginines in renal disease. Nat. Rev. Nephrol. 2011, 7, 275–285. [Google Scholar] [CrossRef]
- Said, M.Y.; Bollenbach, A.; Minovic, I.; van Londen, M.; Frenay, A.R.; de Borst, M.H.; van den Berg, E.; Kayacelebi, A.A.; Tsikas, D.; van Goor, H.; et al. Plasma ADMA, urinary ADMA excretion, and late mortality in renal transplant recipients. Amino Acids 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, H.J.; Jang, H.B.; Kim, H.J.; Ban, H.J.; Kim, K.Y.; Nam, M.S.; Choi, J.S.; Lee, K.T.; Cho, S.B.; et al. Asymmetric dimethylarginine (ADMA) is identified as a potential biomarker of insulin resistance in skeletal muscle. Sci. Rep. 2018, 8, 2133. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.B.; Wang, Y.S.; Luo, Z.F.; Lu, X.Y. SZSJ protects against insomnia by a decrease in ADMA level and an improvement in DDAH production in sleep-deprived rats. Life Sci. 2018, 209, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Zinellu, A.; Fois, A.G.; Mangoni, A.A.; Paliogiannis, P.; Sotgiu, E.; Zinellu, E.; Marras, V.; Pirina, P.; Carru, C. Systemic concentrations of asymmetric dimethylarginine (ADMA) in chronic obstructive pulmonary disease (COPD): State of the art. Amino Acids 2018, 50, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Uzar, E.; Evliyaoglu, O.; Toprak, G.; Acar, A.; Yucel, Y.; Calisir, T.; Cevik, M.U.; Tasdemir, N. Increased asymmetric dimethylarginine and nitric oxide levels in patients with migraine. J. Headache Pain 2011, 12, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, T.; Luo, Z.; Palm, F.; Wilcox, C.S. Cellular ADMA: Regulation and action. Pharmacol. Res. 2009, 60, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar] [CrossRef]
- Masuda, H.; Goto, M.; Tamaoki, S.; Azuma, H. Accelerated intimal hyperplasia and increased endogenous inhibitors for NO synthesis in rabbits with alloxan-induced hyperglycaemia. Br. J. Pharmacol. 1999, 126, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Kaniskan, H.U.; Konze, K.D.; Jin, J. Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58, 1596–1629. [Google Scholar] [CrossRef]
- Hu, H.; Qian, K.; Ho, M.C.; Zheng, Y.G. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin. Investig. Drugs 2016, 25, 335–358. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, Y.G. SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases. ACS Chem. Biol. 2016, 11, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Bayen, S.; Saini, S.; Gaur, P.; Duraisamy, A.J.; Kumar Sharma, A.; Pal, K.; Vats, P.; Singh, S.B. PRMT1 promotes hyperglycemia in a FoxO1-dependent manner, affecting glucose metabolism, during hypobaric hypoxia exposure, in rat model. Endocrine 2018, 59, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Cardounel, A.J.; Zweier, J.L. Endogenous methylarginines regulate neuronal nitric-oxide synthase and prevent excitotoxic injury. J. Biol. Chem. 2002, 277, 33995–34002. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.; Teerlink, T. Plasma concentrations of arginine and asymmetric dimethylarginine do not reflect their intracellular concentrations in peripheral blood mononuclear cells. Metabolism 2013, 62, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Strijbis, K.; Vaz, F.M.; Distel, B. Enzymology of the carnitine biosynthesis pathway. IUBMB Life 2010, 62, 357–362. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fulton, M.D.; Brown, T.; Zheng, Y.G. The Biological Axis of Protein Arginine Methylation and Asymmetric Dimethylarginine. Int. J. Mol. Sci. 2019, 20, 3322. https://doi.org/10.3390/ijms20133322
Fulton MD, Brown T, Zheng YG. The Biological Axis of Protein Arginine Methylation and Asymmetric Dimethylarginine. International Journal of Molecular Sciences. 2019; 20(13):3322. https://doi.org/10.3390/ijms20133322
Chicago/Turabian StyleFulton, Melody D., Tyler Brown, and Y. George Zheng. 2019. "The Biological Axis of Protein Arginine Methylation and Asymmetric Dimethylarginine" International Journal of Molecular Sciences 20, no. 13: 3322. https://doi.org/10.3390/ijms20133322