Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics


Abstract
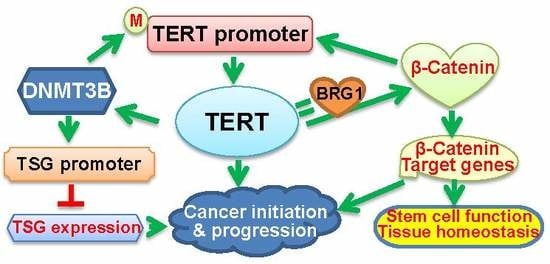
:
1. Introduction
2. The Function of Telomerase Reverse Transcriptase (TERT) in Cancer-Specific DNA Hypermethylation
2.1. The Aberrant DNA Methylation and DNA Methyltransferase (DNMT) Expression in Cancer
2.2. TERT Activation of DNMT3B Transcription in Cancer
3. The Function of TERT in Chromatin Remodeling and Gene Transcription
3.1. Nucleosomes, the switching defective/sucrose non-fermenting (SWI/SNF) Complex, and <ATP-Dependent Chromatin Remodeling
3.2. TERT Interaction withBrahma-Related Gene 1 (BRG)1 to Regulate the Transcription of β-Catenin Target Genes
3.3. The Positive Feedback Loop Between TERT and TERT-Mediated Epigenetic Alterations
3.4. Clinical Implications of TERT Interaction with Epigenetics in Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BMMSC | Bone marrow mesenchymal stem cell |
| CSC | Cancer stem cell |
| DNMT | DNA methyltransferase |
| GIC | Gastrointestinal cancer |
| HCC | Hepatocellular carcinoma |
| MTSS1 | metastasis suppressor 1 |
| NS | nucleostemin |
| RdRP | RNA-dependent RNA polymerase |
| SWI/SNF | Switching defective/sucrose non-fermenting |
| TERC | Telomerase RNA template component |
| TERT | Telomerase reverse transcriptase |
| TF | Transcription factor |
| TGF | Transforming growth factor |
| VEGF | Vascular endothelial growth factor |
References
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yuan, X.; Xu, D. Cancer-Specific Telomerase Reverse Transcriptase (TERT) Promoter Mutations: Biological and Clinical Implications. Genes 2016, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019. [Google Scholar] [CrossRef]
- Chang, W.F.; Wu, Y.H.; Xu, J.; Sung, L.Y. Compromised Chondrocyte Differentiation Capacity in TERC Knockout Mouse Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer. Int. J. Mol. Sci. 2019, 20, 1236. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Zheng, C.; Xu, D. Telomerase as a “stemness” enzyme. Sci. China Life Sci. 2014, 57, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Armando, R.G.; Mengual Gomez, D.L.; Maggio, J.; Sanmartin, M.C.; Gomez, D.E. Telomeropathies: Etiology diagnosis treatment follow-up. Ethical and legal considerations. Clin. Genet. 2019, 96, 3–16. [Google Scholar] [CrossRef]
- Vasko, T.; Kaifie, A.; Stope, M.B.; Kraus, T.; Ziegler, P. Telomeres and Telomerase in Hematopoietic Dysfunction: Prognostic Implications and Pharmacological Interventions. Int. J. Mol. Sci. 2017, 18, 2267. [Google Scholar] [CrossRef] [PubMed]
- Park, J.I.; Venteicher, A.S.; Hong, J.Y.; Choi, J.; Jun, S.; Shkreli, M.; Chang, W.; Meng, Z.; Cheung, P.; Ji, H.; et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature 2009, 460, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Nascimento, E.M.; Gajera, C.R.; Chen, L.; Neuhofer, P.; Garbuzov, A.; Wang, S.; Artandi, S.E. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 2018, 556, 244–248. [Google Scholar] [CrossRef]
- Bar, C.; Povedano, J.M.; Serrano, R.; Benitez-Buelga, C.; Popkes, M.; Formentini, I.; Bobadilla, M.; Bosch, F.; Blasco, M.A. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood 2016, 127, 1770–1779. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Kronstrom, M.; Hellenius, M.L.; Cederholm, T.; Xu, D.; Sjogren, P. Longitudinal changes in leukocyte telomere length and mortality in elderly Swedish men. Aging 2018, 10, 3005–3016. [Google Scholar] [CrossRef] [PubMed]
- Young, A.J. The role of telomeres in the mechanisms and evolution of life-history trade-offs and ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Chen, Y.Y.; Yeh, Y.L.; Wang, Y.J.; Chen, R.J. Stilbene Compounds Inhibit Tumor Growth by the Induction of Cellular Senescence the Inhibition of Telomerase, Activity. Int. J. Mol. Sci. 2019, 20, 2716. [Google Scholar] [CrossRef] [PubMed]
- Seluanov, A.; Gladyshev, V.N.; Vijg, J.; Gorbunova, V. Mechanisms of cancer resistance in long-lived mammals. Nat. Rev. Cancer 2018, 18, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere Length Maintenance in Cancer: At the Crossroad between Telomerase and Alternative Lengthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef] [PubMed]
- Takakura, M.; Kyo, S.; Kanaya, T.; Hirano, H.; Takeda, J.; Yutsudo, M.; Inoue, M. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999, 59, 551–557. [Google Scholar]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/beta-catenin signaling regulates telomerase in stem cells cancer, cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef]
- Zhang, Y.; Toh, L.; Lau, P.; Wang, X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/beta-catenin pathway in human cancer. J. Biol. Chem. 2012, 287, 32494–32511. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Ackermann, S.; Cartolano, M.; Hero, B.; Welte, A.; Kahlert, Y.; Roderwieser, A.; Bartenhagen, C.; Walter, E.; Gecht, J.; Kerschke, L.; et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science 2018, 362, 1165–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentijn, L.J.; Koster, J.; Zwijnenburg, D.A.; Hasselt, N.E.; van Sluis, P.; Volckmann, R.; van Noesel, M.M.; George, R.E.; Tytgat, G.A.; Molenaar, J.J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kost, J.; Sulovari, A.; Wong, N.; Liang, W.; Cao, J.; Li, D. A virome-wide clonal integration analysis platform for discovering cancer viral etiology. Genome Res. 2019, 29, 819–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Kramer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Mu, N.; Wang, N.; Straat, K.; Sofiadis, A.; Guo, Y.; Stenman, A.; Li, K.; Cheng, G.; Zhang, L.; et al. GABPA inhibits invasion/metastasis in papillary thyroid carcinoma by regulating DICER1 expression. Oncogene 2019, 38, 965–979. [Google Scholar] [CrossRef]
- Lee, D.D.; Leao, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolonio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, T.; Maida, Y.; Tomaru, Y.; Yasukawa, M.; Ando, Y.; Kojima, M.; Kasim, V.; Simon, C.; Daub, C.O.; Carninci, P.; et al. Telomerase reverse transcriptase regulates microRNAs. Int. J. Mol. Sci. 2015, 16, 1192–1208. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef]
- Im, E.; Yoon, J.B.; Lee, H.W.; Chung, K.C. Human Telomerase Reverse Transcriptase (hTERT) Positively Regulates 26S Proteasome Activity. J. Cell Physiol. 2017, 232, 2083–2093. [Google Scholar] [CrossRef]
- Hu, C.; Ni, Z.; Li, B.S.; Yong, X.; Yang, X.; Zhang, J.W.; Zhang, D.; Qin, Y.; Jie, M.M.; Dong, H.; et al. hTERT promotes the invasion of gastric cancer cells by enhancing FOXO3a ubiquitination and subsequent ITGB1 upregulation. Gut 2017, 66, 31–42. [Google Scholar] [CrossRef]
- Saretzki, G. Extra-telomeric functions of human telomerase: Cancer, mitochondria and oxidative stress. Curr. Pharm Des. 2014, 20, 6386–6403. [Google Scholar] [CrossRef] [PubMed]
- Masutomi, K.; Possemato, R.; Wong, J.M.; Currier, J.L.; Tothova, Z.; Manola, J.B.; Ganesan, S.; Lansdorp, P.M.; Collins, K.; Hahn, W.C. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc. Natl. Acad. Sci. USA 2005, 102, 8222–8227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Bjorkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Guo, Y.; Wang, X.; Zhao, H.; Ji, Z.; Cheng, C.; Li, L.; Fang, Y.; Xu, D.; Zhu, H.H.; et al. WNT/beta-Catenin Directs Self-Renewal Symmetric Cell Division of hTERT(high) Prostate Cancer Stem Cells. Cancer Res. 2017, 77, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Xi, P.; Zhou, J.; Wang, M.; Cong, Y.S. Human telomerase reverse transcriptase regulates MMP expression independently of telomerase activity via NF-kappaB-dependent transcription. FASEB J. 2013, 27, 4375–4383. [Google Scholar] [CrossRef] [PubMed]
- Green, P.D.; Sharma, N.K.; Santos, J.H. Telomerase Impinges on the Cellular Response to Oxidative Stress Through Mitochondrial ROS-Mediated Regulation of Autophagy. Int. J. Mol. Sci. 2019, 20, 1509. [Google Scholar] [CrossRef]
- Yu, J.; Yuan, X.; Sjoholm, L.; Liu, T.; Kong, F.; Ekstrom, T.J.; Bjorkholm, M.; Xu, D. Telomerase reverse transcriptase regulates DNMT3B expression/aberrant DNA methylation phenotype and AKT activation in hepatocellular carcinoma. Cancer Lett 2018, 434, 33–41. [Google Scholar] [CrossRef]
- Zhang, X.; Li, B.; de Jonge, N.; Bjorkholm, M.; Xu, D. The DNA methylation inhibitor induces telomere dysfunction and apoptosis of leukemia cells that is attenuated by telomerase over-expression. Oncotarget 2015, 6, 4888–4900. [Google Scholar] [CrossRef]
- Ci, X.; Li, B.; Ma, X.; Kong, F.; Zheng, C.; Bjorkholm, M.; Jia, J.; Xu, D. Bortezomib-mediated down-regulation of telomerase and disruption of telomere homeostasis contributes to apoptosis of malignant cells. Oncotarget 2015, 6, 38079–38092. [Google Scholar] [CrossRef] [Green Version]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Antequera, F.; Bird, A. Number of CpG islands and genes in human and mouse. Proc. Natl. Acad. Sci. USA 1993, 90, 11995–11999. [Google Scholar] [CrossRef] [PubMed]
- Stevens, M.; Cheng, J.B.; Li, D.; Xie, M.; Hong, C.; Maire, C.L.; Ligon, K.L.; Hirst, M.; Marra, M.A.; Costello, J.F.; et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res. 2013, 23, 1541–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatada, I.; Fukasawa, M.; Kimura, M.; Morita, S.; Yamada, K.; Yoshikawa, T.; Yamanaka, S.; Endo, C.; Sakurada, A.; Sato, M.; et al. Genome-wide profiling of promoter methylation in human. Oncogene 2006, 25, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Han, T.S.; Ban, H.S.; Hur, K.; Cho, H.S. The Epigenetic Regulation of HCC Metastasis. Int. J. Mol. Sci. 2018, 19, 3978. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, A. Aberrant DNA methylation in hepatocellular carcinoma tumor suppression (Review). Oncol. Lett. 2014, 8, 963–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, D.; Thombre, R.; Dhar, A.; Anant, S. DNA methyltransferases: A novel target for prevention and therapy. Front. Oncol. 2014, 4, 80. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Strazzullo, M.; Matarazzo, M.R. DNMT3B Functions: Novel Insights From Human Disease. Front. Cell Dev. Biol. 2018, 6, 140. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Disc. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Liu, N.; Ding, D.; Hao, W.; Yang, F.; Wu, X.; Wang, M.; Xu, X.; Ju, Z.; Liu, J.P.; Song, Z.; et al. hTERT promotes tumor angiogenesis by activating VEGF via interactions with the Sp1 transcription factor. Nucleic Acids Res. 2016, 44, 8693–8703. [Google Scholar] [CrossRef] [Green Version]
- Jinawath, A.; Miyake, S.; Yanagisawa, Y.; Akiyama, Y.; Yuasa, Y. Transcriptional regulation of the human DNA methyltransferase 3A and 3B genes by Sp3 and Sp1 zinc finger proteins. Biochem. J. 2005, 385, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golob-Schwarzl, N.; Krassnig, S.; Toeglhofer, A.M.; Park, Y.N.; Gogg-Kamerer, M.; Vierlinger, K.; Schroder, F.; Rhee, H.; Schicho, R.; Fickert, P.; et al. New liver cancer biomarkers: PI3K/AKT/mTOR pathway members and eukaryotic translation initiation factors. Eur. J. Cancer 2017, 83, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, B.; Yu, J.; Dahlstrom, J.; Tran, A.N.; Bjorkhom, M.; Xu, D. MYC-dependent downregulation of telomerase by FLT3 inhibitors is required for their therapeutic efficacy on acute myeloid leukemia. Ann. Hematol. 2018, 97, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.Y.; Fu, T.M.; Chen, H.; Ishikawa, T.; Chiang, S.Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef] [PubMed]
- Hamatsu, T.; Rikimaru, T.; Yamashita, Y.; Aishima, S.; Tanaka, S.; Shirabe, K.; Shimada, M.; Toh, Y.; Sugimachi, K. The role of MTA1 gene expression in human hepatocellular carcinoma. Oncol. Rep. 2003, 10, 599–604. [Google Scholar]
- Fan, H.; Chen, L.; Zhang, F.; Quan, Y.; Su, X.; Qiu, X.; Zhao, Z.; Kong, K.L.; Dong, S.; Song, Y.; et al. MTSS1, a novel target of DNA methyltransferase 3B, functions as a tumor suppressor in hepatocellular carcinoma. Oncogene 2012, 31, 2298–2308. [Google Scholar] [CrossRef]
- Fan, H.C.; Chen, C.M.; Chi, C.S.; Tsai, J.D.; Chiang, K.L.; Chang, Y.K.; Lin, S.Z.; Harn, H.J. Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma. Int. J. Mol. Sci. 2019, 20, 200. [Google Scholar] [CrossRef]
- St Pierre, R.; Kadoch, C. Mammalian SWI/SNF complexes in cancer: Emerging therapeutic opportunities. Curr Opin Genet. Dev. 2017, 42, 56–67. [Google Scholar] [CrossRef]
- Kumar, R.; Li, D.Q.; Muller, S.; Knapp, S. Epigenomic regulation of oncogenesis by chromatin remodeling. Oncogene 2016, 35, 4423–4436. [Google Scholar] [CrossRef]
- Savas, S.; Skardasi, G. The SWI/SNF complex subunit genes: Their functions, variations, and links to risk and survival outcomes in human cancers. Crit. Rev. Oncol. Hematol. 2018, 123, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Hurlstone, A.; Musisi, H.; Miles, A.; Bienz, M.; Clevers, H. The chromatin remodelling factor Brg-1 interacts with beta-catenin to promote target gene activation. EMBO J. 2001, 20, 4935–4943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, I.; Cayuela, M.L.; Blasco, M.A. Effects of telomerase and telomere length on epidermal stem cell behavior. Science 2005, 309, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Sarin, K.Y.; Cheung, P.; Gilison, D.; Lee, E.; Tennen, R.I.; Wang, E.; Artandi, M.K.; Oro, A.E.; Artandi, S.E. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature 2005, 436, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Akiyama, K.; Yamaza, T.; You, Y.O.; Xu, X.; Li, B.; Zhao, Y.; Shi, S. Telomerase governs immunomodulatory properties of mesenchymal stem cells by regulating FAS ligand expression. EMBO Mol. Med. 2014, 6, 322–334. [Google Scholar] [PubMed]
- Okamoto, N.; Yasukawa, M.; Nguyen, C.; Kasim, V.; Maida, Y.; Possemato, R.; Shibata, T.; Ligon, K.L.; Fukami, K.; Hahn, W.C.; et al. Maintenance of tumor initiating cells of defined genetic composition by nucleostemin. Proc. Natl. Acad. Sci. USA 2011, 108, 20388–20393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drevytska, T.I.; Nagibin, V.S.; Gurianova, V.L.; Kedlyan, V.R.; Moibenko, A.A.; Dosenko, V.E. Silencing of TERT decreases levels of miR-1, miR-21, miR-29a and miR-208a in cardiomyocytes. Cell Biochem. Funct. 2014, 32, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Cardenas, S.L.; Greider, C.W. Comparing effects of mTR and mTERT deletion on gene expression and DNA damage response: A critical examination of telomere length maintenance-independent roles of telomerase. Nucleic Acids Res. 2010, 38, 60–71. [Google Scholar] [CrossRef]
- Listerman, I.; Gazzaniga, F.S.; Blackburn, E.H. An investigation of the effects of the core protein telomerase reverse transcriptase on Wnt signaling in breast cancer cells. Mol. Cell Biol. 2014, 34, 280–289. [Google Scholar] [CrossRef]
- Wu, Q.; Lian, J.B.; Stein, J.L.; Stein, G.S.; Nickerson, J.A.; Imbalzano, A.N. The BRG1 ATPase of human SWI/SNF chromatin remodeling enzymes as a driver of cancer. Epigenomics 2017, 9, 919–931. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.Q.; Huang, C.; He, X.; Tian, Y.Y.; Zhou, D.X.; He, Y.; Liu, X.H.; Li, J. Feedback regulation of telomerase reverse transcriptase: New insight into the evolving field of telomerase in cancer. Cell Signal. 2013, 25, 2462–2468. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, C.; Fotouhi, O.; Fan, Y.; Wang, K.; Xia, C.; Shi, B.; Zhang, G.; Wang, K.; Kong, F.; et al. TERT Promoter Hypermethylation in Gastrointestinal Cancer: A Potential Stool Biomarker. Oncologist 2017, 22, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Xu, D.; Sofiadis, A.; Hoog, A.; Vukojevic, V.; Backdahl, M.; Zedenius, J.; Larsson, C. Telomerase-dependent and independent telomere maintenance and its clinical implications in medullary thyroid carcinoma. J. Clin. Endocrinol Metab. 2014, 99, E1571–E1579. [Google Scholar] [CrossRef] [PubMed]
- Svahn, F.; Juhlin, C.C.; Paulsson, J.O.; Fotouhi, O.; Zedenius, J.; Larsson, C.; Stenman, A. Telomerase reverse transcriptase promoter hypermethylation is associated with metastatic disease in abdominal paraganglioma. Clin. Endocrinol. (Oxf) 2018, 88, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Hideshima, T.; Hayashi, T.; Tai, Y.T.; Mitsiades, C.S.; Mitsiades, N.; Chauhan, D.; Richardson, P.; Munshi, N.C.; Anderson, K.C. Cytokines modulate telomerase activity in a human multiple myeloma cell line. Cancer Res. 2002, 62, 3876–3882. [Google Scholar] [PubMed]
- Haendeler, J.; Hoffmann, J.; Rahman, S.; Zeiher, A.M.; Dimmeler, S. Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett. 2003, 536, 180–186. [Google Scholar] [CrossRef]
- Kang, S.S.; Kwon, T.; Kwon, D.Y.; Do, S.I. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J. Biol. Chem. 1999, 274, 13085–13090. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Watanabe, H.; Yamamichi, N.; Kondo, S.; Tando, T.; Haraguchi, T.; Mizutani, T.; Sakurai, K.; Fujita, S.; Izumi, T.; et al. Brm transactivates the telomerase reverse transcriptase (TERT) gene and modulates the splicing patterns of its transcripts in concert with p54(nrb). Biochem. J. 2008, 411, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Bougel, S.; Lhermitte, B.; Gallagher, G.; de Flaugergues, J.C.; Janzer, R.C.; Benhattar, J. Methylation of the hTERT promoter: A novel cancer biomarker for leptomeningeal metastasis detection in cerebrospinal fluids. Clin. Cancer Res. 2013, 19, 2216–2223. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, P.; Choufani, S.; Mack, S.; Gallagher, D.; Zhang, C.; Lipman, T.; Zhukova, N.; Walker, E.J.; Martin, D.; Merino, D.; et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: An integrative genomic and molecular study. Lancet Oncol. 2013, 14, 534–542. [Google Scholar] [CrossRef]



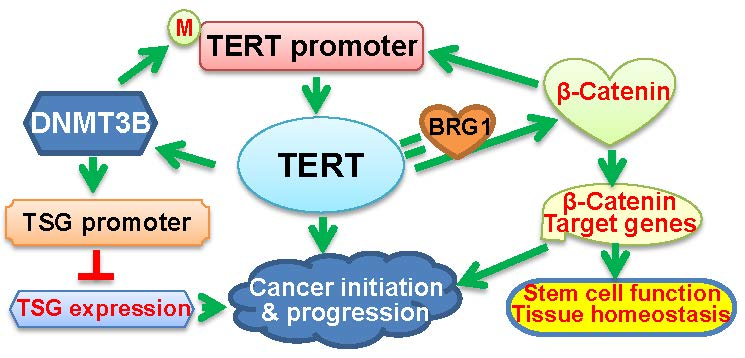{kind=link}
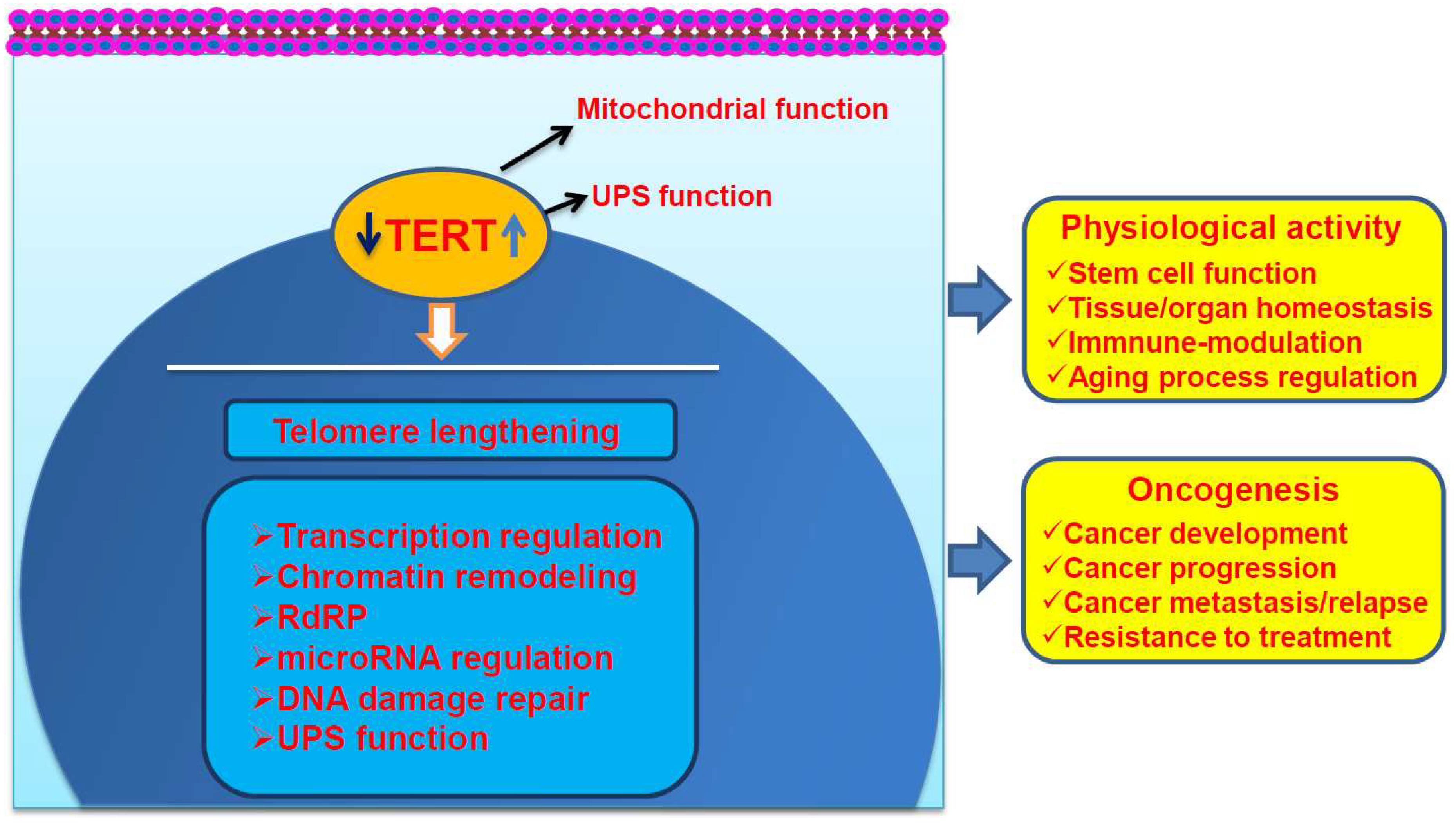{kind=link}
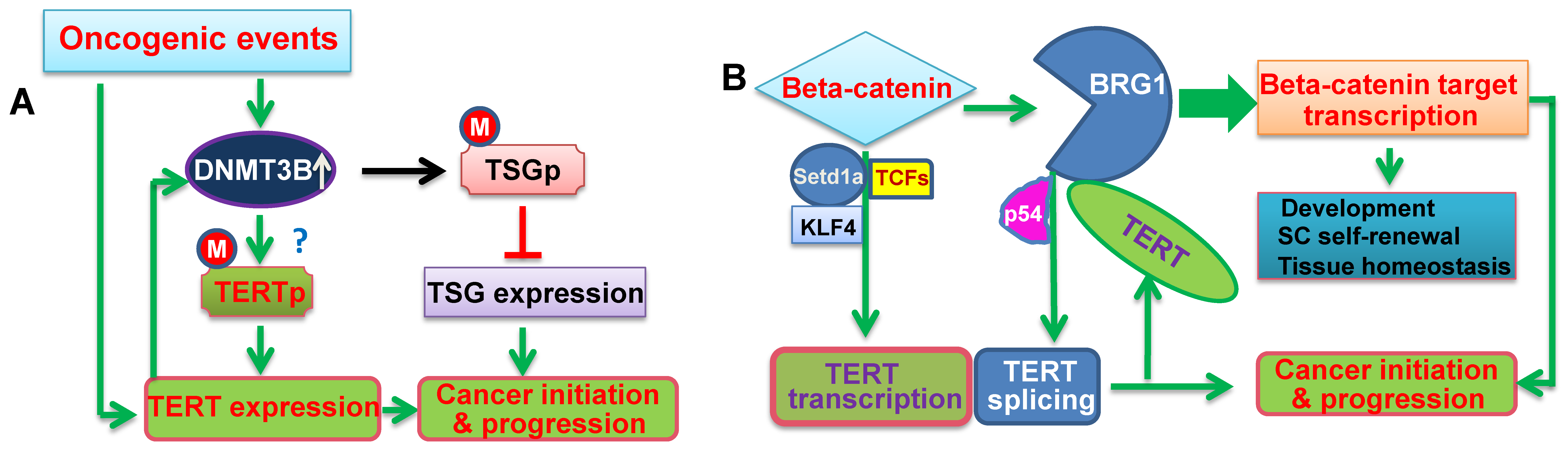{kind=link}
| Cancer Type | N | Spearman Correlation | p Value |
|---|---|---|---|
| Hepatocellular carcinoma | 360 | 0.25 | 1.254 × 10−6 |
| Breast invasive carcinoma | 994 | 0.33 | 6.38 × 10−26 |
| Lung adenocarcinoma | 507 | 0.46 | 1.38 × 10−27 |
| Bladder cancer | 412 | 0.20 | 3.300 × 10−5 |
| Glioblastoma | 273 | 0.49 | 1.34 × 10−9 |
| Cutaneous melanoma | 287 | 0.28 | 1.36 × 10−6 |
| Colorectal adenocarcinoma | 524 | 0.21 | 1.02 × 10−6 |
| Prostate adenocarcinoma | 491 | 0.22 | 1.087 × 10−6 |
| Renal clear cell carcinoma | 446 | 0.28 | 1.29 × 10−9 |
| Pediatric neuroblastoma | 1076 | 0.25 | 3.125 × 10−3 |
| Endometrial carcinoma | 507 | 0.30 | 4.17 × 10−12 |
| Acute myeloid leukemia | 165 | 0.317 | 3.29 × 10−4 |
| Testicular germ cell tumors | 144 | 0.748 | 1.43 × 10−24 |
| Thymoma | 119 | 0.566 | 1.90 × 10−10 |
| Cervical squamous cell carcinoma | 275 | 0.141 | 0.019 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, X.; Xu, D. Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics. Int. J. Mol. Sci. 2019, 20, 3338. https://doi.org/10.3390/ijms20133338
Yuan X, Xu D. Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics. International Journal of Molecular Sciences. 2019; 20(13):3338. https://doi.org/10.3390/ijms20133338
Chicago/Turabian StyleYuan, Xiaotian, and Dawei Xu. 2019. "Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics" International Journal of Molecular Sciences 20, no. 13: 3338. https://doi.org/10.3390/ijms20133338
APA StyleYuan, X., & Xu, D. (2019). Telomerase Reverse Transcriptase (TERT) in Action: Cross-Talking with Epigenetics. International Journal of Molecular Sciences, 20(13), 3338. https://doi.org/10.3390/ijms20133338