Platinum Salts in Patients with Breast Cancer: A Focus on Predictive Factors


 ,
,  and
and 
Abstract
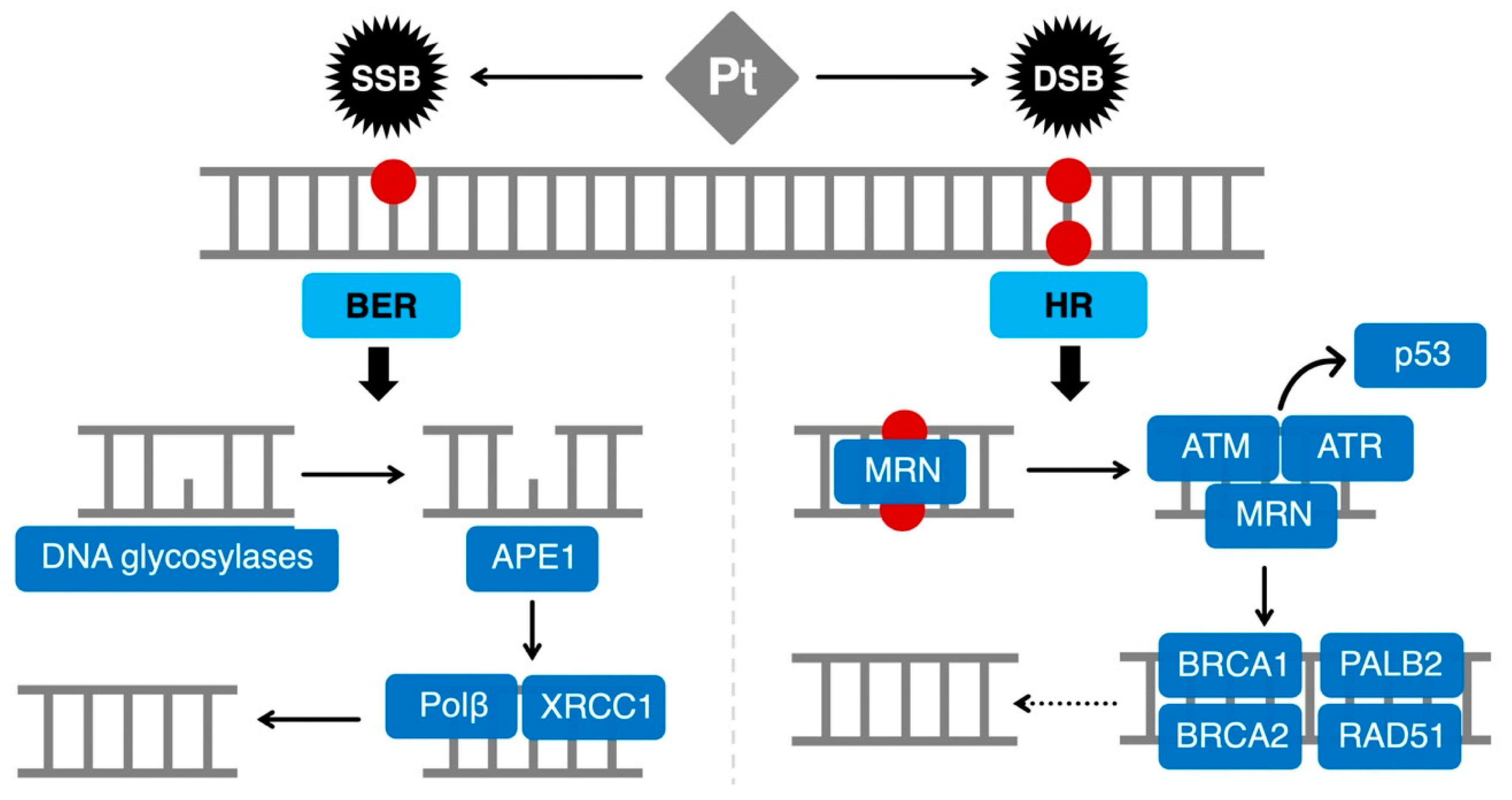
:1. Introduction
2. HRD as a Biomarker of Platinum Sensitivity
2.1. The HR Pathway
2.2. BRCA1/2 Mutations
2.3. BRCAness
3. New Emerging Biomarkers
3.1. Gene Signatures
3.2. p53 Family
3.3. BER
4. Conclusions
Funding
Conflicts of Interest
Abbreviations
| BC | Breast cancer |
| mBC | Metastatic breast cancer |
| TNBC | Triple-negative breast cancer |
| CBDCA | Carboplatin |
| CDDP | Cisplatin |
| SLC31A1 | High Affinity Copper Uptake Protein 1 |
| ATP7B | Copper-Transporting P-type Adenosine Triphosphate |
| ICL | Inter-DNA cross-link |
| SSB | Single-strand break |
| DSB | Double-strand break |
| HR | Homologous recombination repair |
| BER | Base excision repair |
| HRD | Homologous recombination deficiency |
| NHEJ | Non-homologous end joining |
| BRCA1 | Breast Cancer Type 1 Susceptibility Protein |
| BRCA2 | Breast Cancer Type 2 Susceptibility Protein |
| ORR | Objective response rate |
| OS | Overall survival |
| PFS | Progression-free survival |
| PARP | Poly(ADP-ribose) polymerase |
| pCR | Pathologic complete response |
| EFS | Event-free survival |
| DFS | Disease-free survival |
| RCB | Residual cancer burden |
| NPM1 | Nucleophosmin |
| OGG1 | 8-Oxoguanine DNA glycosylase |
| UNG | Uracil-DNA-glyucosylase |
| Polβ | Polymerase β |
| ROS | Reactive oxygen species |
| 8oxoG | Oxidized guanine |
| EFI | Event-free survival |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment. JAMA 2019, 321, 288. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Green, A.R. Molecular classification of breast cancer: What the pathologist needs to know. Pathology 2017, 49, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerratana, L.; Fanotto, V.; Pelizzari, G.; Agostinetto, E.; Puglisi, F. Do platinum salts fit all triple negative breast cancers? Cancer Treat. Rev. 2016, 48, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Poggio, F.; Bruzzone, M.; Ceppi, M.; Pondé, N.F.; La Valle, G.; Del Mastro, L.; de Azambuja, E.; Lambertini, M. Platinum-based neoadjuvant chemotherapy in triple-negative breast cancer: A systematic review and meta-analysis. Ann. Oncol. 2018, 29, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Barni, S.; Bregni, G.; de Braud, F.; Di Cosimo, S. Platinum salts in advanced breast cancer: A systematic review and meta-analysis of randomized clinical trials. Breast Cancer Res. Treat. 2016, 160, 425–437. [Google Scholar] [CrossRef]
- Rocha, C.; Silva, M.; Quinet, A.; Cabral-Neto, J.; Menck, C. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Zwelling, L.A.; Kohn, K.W. Effects of cisplatin on dna and the possible relationships to cytotoxicity and mutagenicity in mammalian cells. Cisplatin 2013, 73, 21–35. [Google Scholar]
- Rosenberg, B. Cisplatin: Its history and possible mechanisms of action. In Cisplatin; Academic Press: New York, NY, USA, 2013; pp. 9–20. [Google Scholar]
- Bradner, W.T.; Rose, W.C.; Huftalen, J.B. antitumor activity of platinum analogs. In Cisplatin; Academic Press: New York, NY, USA, 2013; pp. 171–182. [Google Scholar]
- Cui, Y.; König, J.; Buchholz, J.K.; Spring, H.; Leier, I.; Keppler, D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharmacol. 1999, 55, 929–937. [Google Scholar] [PubMed]
- Komatsu, M.; Sumizawa, T.; Mutoh, M.; Chen, Z.S.; Terada, K.; Furukawa, T.; Yang, X.L.; Gao, H.; Miura, N.; Sugiyama, T.; et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000, 60, 1312–1316. [Google Scholar] [PubMed]
- Holzer, A.K.; Samimi, G.; Katano, K.; Naerdemann, W.; Lin, X.; Safaei, R.; Howell, S.B. The Copper Influx Transporter Human Copper Transport Protein 1 Regulates the Uptake of Cisplatin in Human Ovarian Carcinoma Cells. Mol. Pharmacol. 2004, 66, 817–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safaei, R.; Katano, K.; Samimi, G.; Naerdemann, W.; Stevenson, J.L.; Rochdi, M.; Howell, S.B. Cross-resistance to cisplatin in cells with acquired resistance to copper. Cancer Chemother. Pharmacol. 2004, 53, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Katano, K.; Kondo, A.; Safaei, R.; Holzer, A.; Samimi, G.; Mishima, M.; Kuo, Y.-M.; Rochdi, M.; Howell, S.B. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002, 62, 6559–6565. [Google Scholar] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Cross-linking of glutathione to DNA by cancer chemotherapeutic platinum coordination complexes. Chem. Biol. Interact. 1987, 61, 241–248. [Google Scholar] [CrossRef]
- Kasahara, K.; Fujiwara, Y.; Nishio, K.; Ohmori, T.; Sugimoto, Y.; Komiya, K.; Matsuda, T.; Saijo, N. Metallothionein content correlates with the sensitivity of human small cell lung cancer cell lines to cisplatin. Cancer Res. 1991, 51, 3237–3242. [Google Scholar] [PubMed]
- Gonzalez, V.M.; Fuertes, M.A.; Alonso, C.; Perez, J.M. Is Cisplatin-Induced Cell Death Always Produced by Apoptosis? Mol. Pharmacol. 2001, 59, 657–663. [Google Scholar] [CrossRef]
- Becker, J.P.; Weiss, J.; Theile, D. Cisplatin, oxaliplatin, and carboplatin unequally inhibit in vitro mRNA translation. Toxicol. Lett. 2014, 225, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Srivastava, D.K.; Zmudzka, B.Z.; Wilson, S.H. Strategic down-regulation of DNA polymerase β by antisense RNA sensitizes mammalian cells to specific DNA damaging agents. Nucleic Acids Res. 1995, 23, 3810–3815. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.R.; Jiang, Y.; Guo, C.; Reed, A.; Meng, H.; Vasko, M.R. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Guo, C.; Thompson, E.L.; Jiang, Y.; Kelley, M.R.; Vasko, M.R.; Lee, S.H. APE1, the DNA base excision repair protein, regulates the removal of platinum adducts in sensory neuronal cultures by NER. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2015, 779, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothandapani, A.; Dangeti, V.S.M.N.; Brown, A.R.; Banze, L.A.; Wang, X.H.; Sobol, R.W.; Patrick, S.M. Novel role of base excision repair in mediating cisplatin cytotoxicity. J. Biol. Chem. 2011, 286, 14564–14574. [Google Scholar] [CrossRef] [PubMed]
- Kothandapani, A.; Sawant, A.; Dangeti, V.S.M.N.; Sobol, R.W.; Patrick, S.M. Epistatic role of base excision repair and mismatch repair pathways in mediating cisplatin cytotoxicity. Nucleic Acids Res. 2013, 41, 7332–7343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poletto, M.; Lirussi, L.; Wilson, D.M.; Tell, G. Nucleophosmin modulates stability, activity, and nucleolar accumulation of base excision repair proteins. Mol. Biol. Cell 2014, 25, 1641–1652. [Google Scholar] [CrossRef]
- Preston, T.J.; Henderson, J.T.; McCallum, G.P.; Wells, P.G. Base excision repair of reactive oxygen species-initiated 7,8-dihydro-8-oxo-2′-deoxyguanosine inhibits the cytotoxicity of platinum anticancer drugs. Mol. Cancer Ther. 2009, 8, 2015–2026. [Google Scholar] [CrossRef]
- Sawant, A.; Floyd, A.M.; Dangeti, M.; Lei, W.; Sobol, R.W.; Patrick, S.M. Differential role of base excision repair proteins in mediating cisplatin cytotoxicity. DNA Repair 2017, 51, 46–59. [Google Scholar] [CrossRef] [Green Version]
- Slyskova, J.; Sabatella, M.; Ribeiro-Silva, C.; Stok, C.; Theil, A.F.; Vermeulen, W.; Lans, H. Base and nucleotide excision repair facilitate resolution of platinum drugs-induced transcription blockage. Nucleic Acids Res. 2018, 46, 9537–9549. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Parsons, J.; Nicolay, N.H.; Caporali, S.; Harrington, C.F.; Singh, R.; Finch, D.; Datri, S.; Farmer, P.B.; Johnston, P.G.; et al. Cells deficient in the base excision repair protein, DNA polymerase beta, are hypersensitive to oxaliplatin chemotherapy. Oncogene 2010, 29, 463–468. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of Strand Breaks by Homologous Recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Chen, C.-C.; Feng, W.; Lim, P.X.; Kass, E.M.; Jasin, M. Homology-Directed Repair and the Role of BRCA1, BRCA2, and Related Proteins in Genome Integrity and Cancer. Annu. Rev. Cancer Biol. 2018, 2, 313–336. [Google Scholar] [CrossRef]
- Godin, S.K.; Sullivan, M.R.; Bernstein, K.A. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem. Cell Biol. 2016, 94, 407–418. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Klemp, J.R.; Kimler, B.F.; Mahnken, J.D.; Geier, L.J.; Khan, Q.J.; Elia, M.; Connor, C.S.; McGinness, M.K.; Mammen, J.M.W.; et al. Germline BRCA mutation evaluation in a prospective triple-negative breast cancer registry: Implications for hereditary breast and/or ovarian cancer syndrome testing. Breast Cancer Res. Treat. 2014, 145, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Chahwan, C.; Huffman, J.L.; Tainer, J.A. Structure and function of the double-strand break repair machinery. DNA Repair 2004, 3, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Chen, J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef]
- Byrski, T.; Gronwald, J.; Huzarski, T.; Grzybowska, E.; Budryk, M.; Stawicka, M.; Mierzwa, T.; Szwiec, M.; Wiśniowski, R.; Siolek, M.; et al. Pathologic Complete Response Rates in Young Women with BRCA1-Positive Breast Cancers after Neoadjuvant Chemotherapy. J. Clin. Oncol. 2010, 28, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Byrski, T.; Dent, R.; Blecharz, P.; Foszczynska-Kloda, M.; Gronwald, J.; Huzarski, T.; Cybulski, C.; Marczyk, E.; Chrzan, R.; Eisen, A.; et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res. 2012, 14, R110. [Google Scholar] [CrossRef]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J.; Mayer, E.L.; He, L.; Traina, T.A.; Carey, L.A.; Krag, K.J.; Rugo, H.S.; Liu, M.C.; Stearns, V.; Come, S.E.; et al. TBCRC009: A Multicenter Phase II Clinical Trial of Platinum Monotherapy with Biomarker Assessment in Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2015, 33, 1902–1909. [Google Scholar] [CrossRef]
- Yardley, D.A.; Coleman, R.; Conte, P.; Cortes, J.; Brufsky, A.; Shtivelband, M.; Young, R.; Bengala, C.; Ali, H.; Eakel, J.; et al. nab-Paclitaxel plus carboplatin or gemcitabine versus gemcitabine plus carboplatin as first-line treatment of patients with triple-negative metastatic breast cancer: Results from the tnAcity trial. Ann. Oncol. 2018, 29, 1763–1770. [Google Scholar] [CrossRef]
- Sikov, W.M.; Berry, D.A.; Perou, C.M.; Singh, B.; Cirrincione, C.T.; Tolaney, S.M.; Kuzma, C.S.; Pluard, T.J.; Somlo, G.; Port, E.R.; et al. Impact of the Addition of Carboplatin and/or Bevacizumab to Neoadjuvant Once-per-Week Paclitaxel Followed by Dose-Dense Doxorubicin and Cyclophosphamide on Pathologic Complete Response Rates in Stage II to III Triple-Negative Breast Cancer: CALGB 40603 (Alliance). J. Clin. Oncol. 2015, 33, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Loibl, S.; Weber, K.E.; Timms, K.M.; Elkin, E.P.; Hahnen, E.; Fasching, P.A.; Lederer, B.; Denkert, C.; Schneeweiss, A.; Braun, S.; et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response—Final results from GeparSixto. Ann. Oncol. 2018, 29, 2341–2347. [Google Scholar] [CrossRef] [PubMed]
- Von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756. [Google Scholar] [CrossRef]
- Sikov, W.; Berry, D.; Perou, C.; Singh, B.; Cirrincione, C.; Tolaney, S.; Somlo, G.; Port, E.; Qamar, R.; Sturtz, K.; et al. Abstract S2-05: Event-free and overall survival following neoadjuvant weekly paclitaxel and dose-dense AC +/-carboplatin and/or bevacizumab in triple-negative breast cancer: Outcomes from CALGB 40603 (Alliance). Cancer Res. 2016, 76, S2-05. [Google Scholar] [CrossRef]
- Hahnen, E.; Lederer, B.; Hauke, J.; Loibl, S.; Kröber, S.; Schneeweiss, A.; Denkert, C.; Fasching, P.A.; Blohmer, J.U.; Jackisch, C.; et al. Germline Mutation Status, Pathological Complete Response, and Disease-Free Survival in Triple-Negative Breast Cancer. JAMA Oncol. 2017, 3, 1378. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; O’Shaughnessy, J.; Untch, M.; Sikov, W.M.; Rugo, H.S.; McKee, M.D.; Huober, J.; Golshan, M.; von Minckwitz, G.; Maag, D.; et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): A randomised, phase 3 trial. Lancet Oncol. 2018, 19, 497–509. [Google Scholar] [CrossRef]
- Telli, M.; McMillan, A.; Ford, J.; Richardson, A.; Silver, D.; Isakoff, S.; Kaklamani, V.; Gradishar, W.; Stearns, V.; Connolly, R.; et al. Abstract P3-07-12: Homologous recombination deficiency (HRD) as a predictive biomarker of response to neoadjuvant platinum-based therapy in patients with triple negative breast cancer (TNBC): A pooled analysis. Cancer Res. 2016, 76, 625–630. [Google Scholar] [CrossRef]
- Kaklamani, V.G.; Jeruss, J.S.; Hughes, E.; Siziopikou, K.; Timms, K.M.; Gutin, A.; Abkevich, V.; Sangale, Z.; Solimeno, C.; Brown, K.L.; et al. Phase II neoadjuvant clinical trial of carboplatin and eribulin in women with triple negative early-stage breast cancer (NCT01372579). Breast Cancer Res. Treat. 2015, 151, 629–638. [Google Scholar] [CrossRef]
- Sharma, P.; Barlow, W.E.; Godwin, A.K.; Pathak, H.; Isakova, K.; Williams, D.; Timms, K.M.; Hartman, A.R.; Wenstrup, R.J.; Linden, H.M.; et al. Impact of homologous recombination deficiency biomarkers on outcomes in patients with triple-negative breast cancer treated with adjuvant doxorubicin and cyclophosphamide (SWOG S9313). Ann. Oncol. 2018, 29, 654–660. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.-O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant Cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- De Summa, S.; Pinto, R.; Sambiasi, D.; Petriella, D.; Paradiso, V.; Paradiso, A.; Tommasi, S. BRCAness: A deeper insight into basal-like breast tumors. Ann. Oncol. 2013, 24, viii13–viii21. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Minckwitz, G.; Timms, K.; Untch, M.; Elkin, E.P.; Fasching, P.A.; Schneeweiss, A.; Salat, C.; Rezai, M.; Blohmer, J.U.; Zahm, D.M.; et al. Prediction of pathological complete response (pCR) by Homologous Recombination Deficiency (HRD) after carboplatin-containing neoadjuvant chemotherapy in patients with TNBC: Results from GeparSixto. J. Clin. Oncol. 2015, 33, 1004. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A.; Kim, D.K.; Song, S.-H.; Kim, H.-J.; Lee, K.-H.; Kim, T.-Y.; Han, S.-W.; Oh, D.-Y.; Kim, T.-Y.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef]
- Jiang, Y.-Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.-D.; Liu, Y.-R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef] [Green Version]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Jumppanen, M.; Gruvberger-Saal, S.; Kauraniemi, P.; Tanner, M.; Bendahl, P.-O.; Lundin, M.; Krogh, M.; Kataja, P.; Borg, Å.; Fernö, M.; et al. Basal-like phenotype is not associated with patient survival in estrogen-receptor-negative breast cancers. Breast Cancer Res. 2007, 9, R16. [Google Scholar] [CrossRef] [PubMed]
- Calza, S.; Hall, P.; Auer, G.; Bjöhle, J.; Klaar, S.; Kronenwett, U.; Liu, E.T.; Miller, L.; Ploner, A.; Smeds, J.; et al. Intrinsic molecular signature of breast cancer in a population-based cohort of 412 patients. Breast Cancer Res. 2006, 8, R34. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Perou, C.M.; Symmans, W.F.; Ibrahim, N.; Cristofanilli, M.; Anderson, K.; Hess, K.R.; Stec, J.; Ayers, M.; Wagner, P.; et al. Breast Cancer Molecular Subtypes Respond Differently to Preoperative Chemotherapy. Clin. Cancer Res. 2005, 11, 5678–5685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertucci, F.; Finetti, P.; Cervera, N.; Esterni, B.; Hermitte, F.; Viens, P.; Birnbaum, D. How basal are triple-negative breast cancers? Int. J. Cancer 2008, 123, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Hubalek, M.; Czech, T.; Müller, H. Biological Subtypes of Triple-Negative Breast Cancer. Breast Care 2017, 12, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, C.; Oh, D.S.; Wessels, L.; Weigelt, B.; Nuyten, D.S.A.; Nobel, A.B.; van’t Veer, L.J.; Perou, C.M. Concordance among Gene-Expression–Based Predictors for Breast Cancer. N. Engl. J. Med. 2006, 355, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.U.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-Like Breast Cancer Defined by Five Biomarkers Has Superior Prognostic Value than Triple-Negative Phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcel, V.; Nguyen Van Long, F.; Diaz, J.-J. 40 Years of Research Put p53 in Translation. Cancers 2018, 10, 152. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Pestell, K.E.; Hobbs, S.M.; Titley, J.C.; Kelland, L.R.; Walton, M.I. Effect of p53 status on sensitivity to platinum complexes in a human ovarian cancer cell line. Mol. Pharmacol. 2000, 57, 503–511. [Google Scholar] [CrossRef]
- Holstege, H.; Joosse, S.A.; van Oostrom, C.T.M.; Nederlof, P.M.; de Vries, A.; Jonkers, J. High Incidence of Protein-Truncating TP53 Mutations in BRCA1-Related Breast Cancer. Cancer Res. 2009, 69, 3625–3633. [Google Scholar] [CrossRef] [PubMed]
- Allocati, N.; Di Ilio, C.; De Laurenzi, V. p63/p73 in the control of cell cycle and cell death. Exp. Cell Res. 2012, 318, 1285–1290. [Google Scholar] [CrossRef]
- Rocco, J.W.; Leong, C.-O.; Kuperwasser, N.; DeYoung, M.P.; Ellisen, L.W. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006, 9, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Leong, C.-O.; Vidnovic, N.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J. Clin. Investig. 2007, 117, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Limpose, K.L.; Corbett, A.H.; Doetsch, P.W. BERing the burden of damage: Pathway crosstalk and posttranslational modification of base excision repair proteins regulate DNA damage management. DNA Repair 2017, 56, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Hübscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- Antoniali, G.; Malfatti, M.C.; Tell, G. Unveiling the non-repair face of the Base Excision Repair pathway in RNA processing: A missing link between DNA repair and gene expression? DNA Repair 2017. [Google Scholar] [CrossRef] [PubMed]
- Allinson, S.L.; Sleeth, K.M.; Matthewman, G.E.; Dianov, G.L. Orchestration of base excision repair by controlling the rates of enzymatic activities. DNA Repair 2004, 3, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Almeida, K.H.; Sobol, R.W. A unified view of base excision repair: Lesion-dependent protein complexes regulated by post-translational modification. DNA Repair 2007, 6, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Déry, U.; Masson, J.-Y. Twists and turns in the function of DNA damage signaling and repair proteins by post-translational modifications. DNA Repair 2007, 6, 561–577. [Google Scholar] [CrossRef]
- Wilson, S.H.; Kunkel, T.A. Passing the baton in base excision repair. Nat. Struct. Biol. 2000, 7, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Wilson, D.M. Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radic. Biol. Med. 2005, 38, 1121–1138. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 Interacts with NPM1 within Nucleoli and Plays a Role in the rRNA Quality Control Process. Mol. Cell. Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faucher, F.; Doubli, S.; Jia, Z. 8-oxoguanine DNA glycosylases: One lesion, three subfamilies. Int. J. Mol. Sci. 2012, 13, 6711–6729. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O.; Mechetin, G.V.; Nevinsky, G.A. Uracil-DNA glycosylase: Structural, thermodynamic and kinetic aspects of lesion search and recognition. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2010, 685, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tell, G.; Lirussi, L.; Antoniali, G.; Wilson III, D.M.; Poletto, M. The Abasic Endonuclease APE1: Much more than a DNA Repair Enzyme. In The Base Excision Repair Pathway; World Scientific: Singapore, 2016; pp. 219–251. [Google Scholar]
- Wilson, D.M. Ape1 abasic endonuclease activity is regulated by magnesium and potassium concentrations and is robust on alternative DNA structures. J. Mol. Biol. 2005, 345, 1003–1014. [Google Scholar] [CrossRef]
- Sobol, R.W.; Horton, J.K.; Kühn, R.; Gu, H.; Singhal, R.K.; Prasad, R.; Rajewsky, K.; Wilson, S.H. Requirement of mammalian DNA polymerase-beta in base-excision repair. Nature 1996, 379, 183–186. [Google Scholar] [CrossRef]
- Sung, J.-S.; DeMott, M.S.; Demple, B. Long-patch base excision DNA repair of 2-deoxyribonolactone prevents the formation of DNA-protein cross-links with DNA polymerase beta. J. Biol. Chem. 2005, 280, 39095–39103. [Google Scholar] [CrossRef]
- Brozovic, A.; Ambriović-Ristov, A.; Osmak, M. The relationship between cisplatin-Induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef]
- Gul, T.; Mehraj Balkhi, H.; Haq, E. Reactive Oxygen Species (ROS). In Evaluation of Cellular Processes by in Vitro Assays; Bentham Science Publishers: Soest, The Netherlands, 2018; pp. 58–67. [Google Scholar]
- Duarte, D.B.; Vasko, M.R. The Role of DNA Damage and Repair in Neurotoxicity Caused by Cancer Therapies. In DNA Repair in Cancer Therapy; Elsevier: Amsterdam, The Netherlands, 2012; pp. 283–299. ISBN 9780123849991. [Google Scholar]
- Kelley, M.R.; Wikel, J.H.; Guo, C.; Pollok, K.E.; Bailey, B.J.; Wireman, R.; Fishel, M.L.; Vasko, M.R. Identification and Characterization of New Chemical Entities Targeting Apurinic/Apyrimidinic Endonuclease 1 for the Prevention of Chemotherapy-Induced Peripheral Neuropathy. J. Pharmacol. Exp. Ther. 2016, 359, 300–309. [Google Scholar] [CrossRef]
- Vasko, M.R.; Guo, C.; Thompson, E.L.; Kelley, M.R. The repair function of the multifunctional DNA repair/redox protein APE1 is neuroprotective after ionizing radiation. DNA Repair 2011, 10, 942–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frossi, B.; Antoniali, G.; Yu, K.; Akhtar, N.; Kaplan, M.H.; Kelley, M.R.; Tell, G.; Pucillo, C.E.M. Endonuclease and redox activities of human apurinic/apyrimidinic endonuclease 1 have distinctive and essential functions in IgA class switch recombination. J. Biol. Chem. 2019, jbc.RA118.006601. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Xiang, D.-B.; Yang, X.-Q.; Chen, L.-S.; Li, M.-X.; Zhong, Z.-Y.; Zhang, Y.-S. APE1 overexpression is associated with cisplatin resistance in non-small cell lung cancer and targeted inhibition of APE1 enhances the activity of cisplatin in A549 cells. Lung Cancer 2009, 66, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, M.C.; Gerratana, L.; Dalla, E.; Isola, M.; Damante, G.; Di Loreto, C.; Puglisi, F.; Tell, G. APE1 and NPM1 protect cancer cells from platinum compounds cytotoxicity and their expression pattern has a prognostic value in TNBC. J. Exp. Clin. Cancer Res. 2019, in press. [Google Scholar]


{kind=link}
| Reference | Setting | Biomarker | Treatments | Outcomes/Observations |
|---|---|---|---|---|
| BRCA1/2 Mutations | ||||
| TNT [49] (phase III) | Stage IV TNBC | BRCA1/2m | CBDCA versus Docetaxel | Increased ORR and PFS with Carboplatin versus Docetaxel |
| TBCRC009 [50] (phase II) | Stage IV TNBC | BRCA1/2m | CBDCA or CDDP | Increased ORR in BRCA1/2m versus BRCA1/2 wt |
| BROCADE [51] (phase II) | Stage IV BC with BRCA1/2m | BRCA1/2m | CP versus CPV versus TV | Increased ORR and PFS with CP and CPV versus TV |
| GeparSixto [53] (phase II) | Stage II–III TNBC | BRCA1/2m | P+A+Bev ± CBDCA | No additive effect on pCR for carboplatin |
| BrighTNess [57] (phase III) | Stage II–III TNBC | BRCA1/2m | CP ± V then AC | No additive effect on pCR for carboplatin |
| BRCAness | ||||
| Telli et al. [58] (pooled analysis) | Localized TNBC | HRD score > 41 | Various platinum containing regimens | Increased pCR in HRD versus HRD < 41 |
| Kaklamani et al. [59] (phase II) | Stage I–III TNBC | HR status (HRD score + BRCA1/2m) | CBDCA + E | HRD status and the HRD score predict pCR |
| GeparSixto [53] (phase II) | Stage II–III TNBC | HRD status * | P+A+Bev ± CBDCA | HRD positive status associated with increased pCR versus HRD negative Adding carboplatin increased pCR in HRD positive but not in HRD negative tumors |
| SWOG9313 [60] (phase III) | Stage I–II TNBC | HRD status * | Concomitant versus sequential AC | HRD positive status associated with DFS. No significative trend observed with OS |
| Gene Signatures and p53 Family | ||||
| Lehmann et al. [61] (in vitro analysis) | TNBC cell lines | BL | Platinum salts | Increased sensitivity |
| TNT [49] (phase III) | Stage IV TNBC | BL, core-basal | CBDCA versus Docetaxel | Reduced ORR and PFS in not-BL and not-core basal |
| Silver et al. [62] | Stage II–III TNBC | p53 NSM | Cisplatin | Increased pCR versus not p53 NSM |
| Silver et al. [62] | Stage II–III TNBC | ∆Np63/TAp73 ratio > 2 | Cisplatin | Numerical increased pCR vs. ∆Np63/TAp73 ratio < 2 |
| BER | ||||
| Kothandapani et al. [26] (in vitro analysis) | MDA-MB-231 TNBC cell line | PolB | CDDP | Upon KO, cells are resistant to CDDP treatment |
| Kothandapani et al. [26] (in vitro analysis) | MDA-MB-231 TNBC cell line | UNG | CDDP | Upon KO, cells are resistant to CDDP treatment |
| Kothandapani et al. [26] (in vitro analysis) | MDA-MB-231 TNBC cell line | APE1 | CDDP | Upon MX, APE1 inhibitor, cells are resistant to CDDP treatment |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garutti, M.; Pelizzari, G.; Bartoletti, M.; Malfatti, M.C.; Gerratana, L.; Tell, G.; Puglisi, F. Platinum Salts in Patients with Breast Cancer: A Focus on Predictive Factors. Int. J. Mol. Sci. 2019, 20, 3390. https://doi.org/10.3390/ijms20143390
Garutti M, Pelizzari G, Bartoletti M, Malfatti MC, Gerratana L, Tell G, Puglisi F. Platinum Salts in Patients with Breast Cancer: A Focus on Predictive Factors. International Journal of Molecular Sciences. 2019; 20(14):3390. https://doi.org/10.3390/ijms20143390
Chicago/Turabian StyleGarutti, Mattia, Giacomo Pelizzari, Michele Bartoletti, Matilde Clarissa Malfatti, Lorenzo Gerratana, Gianluca Tell, and Fabio Puglisi. 2019. "Platinum Salts in Patients with Breast Cancer: A Focus on Predictive Factors" International Journal of Molecular Sciences 20, no. 14: 3390. https://doi.org/10.3390/ijms20143390
APA StyleGarutti, M., Pelizzari, G., Bartoletti, M., Malfatti, M. C., Gerratana, L., Tell, G., & Puglisi, F. (2019). Platinum Salts in Patients with Breast Cancer: A Focus on Predictive Factors. International Journal of Molecular Sciences, 20(14), 3390. https://doi.org/10.3390/ijms20143390