Cerebrovascular and Neurological Disorders: Protective Role of NRF2



Abstract
:
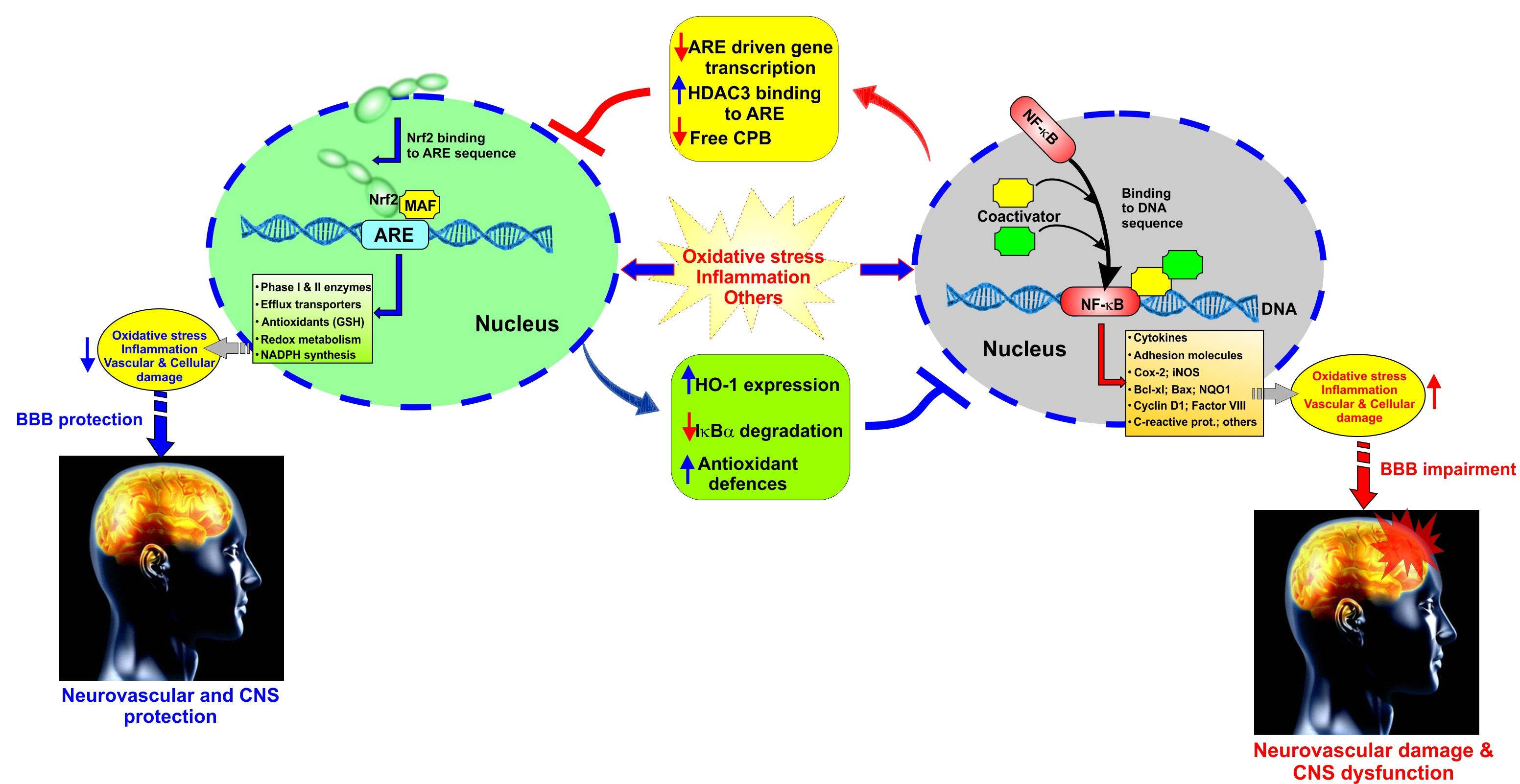{kind=link}
{kind=link}
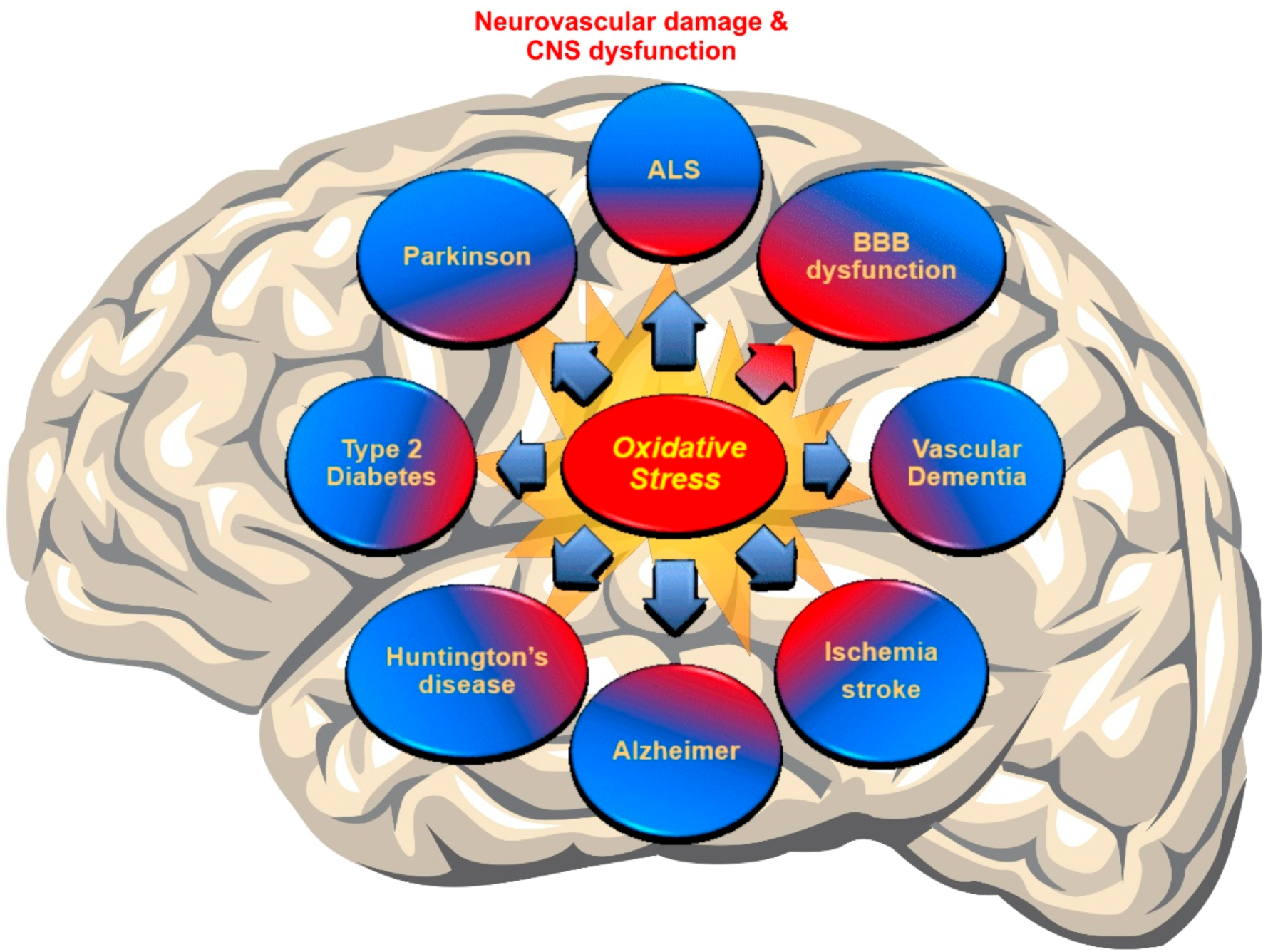{kind=link}
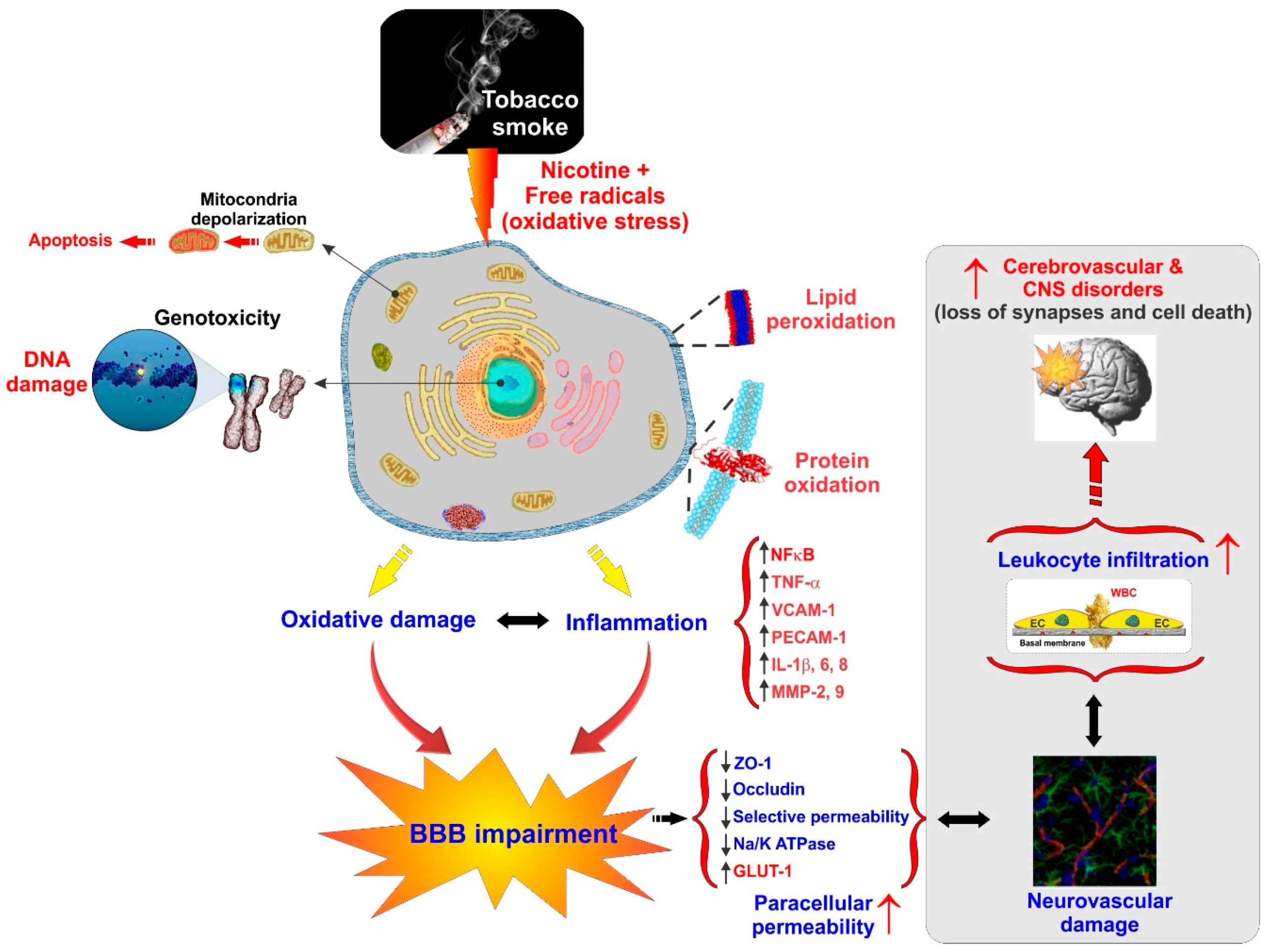{kind=link}
1. Introduction
2. NRF2 Regulation and Response to Oxidative Stress
3. Crosstalk between NRF2 and Other Regulatory Signaling Pathways
4. Role of NRF2 in Aging and Traumatic Brain Injury
5. Ischemic Stroke and Protective Role of NRF2
6. Role of NRF2 in Neurodegenerative Diseases
7. Role of NRF2 in Blood-Brain Barrier (BBB) Integrity and Function
8. Role of NRF2 in Tobacco Smoke-Induced Cerebrovascular Disorders
9. Role of NRF2 in Hyperglycemia
10. NRF2 Enhancers for the Treatment of Cerebrovascular Disorders: Repurposing of Antidiabetic Drugs
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| αSyn | Alpha-synuclein |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | AMP-activated protein kinase |
| ARE | Anti-oxidant response element |
| BBB | Blood–brain barrier |
| BMVEC | Brain microvascular endothelial cells |
| CAT | Catalase |
| CNC | Cap‘n’collar |
| COPD | Chronic obstructive pulmonary disease |
| EGCG | Epigallocatechin gallate |
| Glut 1 | Glucose transporter |
| GSH Px | Glutathione peroxidase |
| HD | Huntington’s disease |
| HO-1 | Heme oxygenase-1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| IKK | IκB kinase |
| KEAP1 | Kelc-like ECH-associated protein 1 |
| MF | Metformin |
| MS | Multiple sclerosis |
| NLRP3 | Nod-like receptor protein 3 |
| NQO-1 | NAD(P)H: quinone reductase I |
| NTF2 | Nuclear factor erythroid 2-related factor |
| PAMPs | Pathogen-associated molecular patterns |
| PD | Parkinson’s disease |
| PGC-1α | Proliferator-activated receptor gamma coactivator 1-alpha |
| PKC | Protein kinase C |
| PPARγ | Peroxisome proliferator-activated receptor gamma |
| PRR | Pattern recognition receptor |
| ROS | Reactive oxygen species |
| RSG | Rosiglitazone |
| SFN | Sulforaphane |
| SOD | Superoxide dismutase |
| TD2M | Type-2 diabetes mellitus |
| TBHQ | Terbutylhydroquinone |
| TBI | Traumatic brain injury |
| TEER | Trans-endothelial electrical resistance |
| TLRs | Toll-like receptors |
| TJ | Tight junction |
| TMCAO | Transient middle artery occlusion |
| TS | Tobacco smoking |
| Trx1 | Thioredoxin-1 |
| ZO-1 | Zonulae occludentes-1 |
References
- Huang, Y.; Li, W.J.; Su, Z.Y.; Kong, A.-N.T. The complexity of the Nrf2 pathway: beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2–ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Kaisar, M.A.; Vijay, V.; Desai, V.G.; Prasad, S.; Cucullo, L. In Vitro Modulation of Redox and Metabolism Interplay at the Brain Vascular Endothelium: Genomic and Proteomic Profiles of Sulforaphane Activity. Sci. Rep. 2018, 8, 12708. [Google Scholar] [CrossRef] [PubMed]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2–ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Patil, J.; D’angelo, B.; Weber, S.G.; Mallard, C. NRF2-regulation in brain health and disease: implication of cerebral inflammation. Neuropharmacology 2014, 79, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1–Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Mishra, M. Epigenetic regulation of redox signaling in diabetic retinopathy: Role of Nrf2. Free Radic. Biol. Med. 2017, 103, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Sajja, R.K.; Park, J.H.; Naik, P.; Kaisar, M.A.; Cucullo, L. Impact of cigarette smoke extract and hyperglycemic conditions on blood–brain barrier endothelial cells. Fluids Barriers CNS 2015, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Tufekci, K.U.; Civi Bayin, E.; Genc, S.; Genc, K. The Nrf2/ARE pathway: A promising target to counteract mitochondrial dysfunction in Parkinson’s disease. Parkinson’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Baird, L.; Zhang, Y.; Hargreaves, I.; Chalasani, A.; Land, J.M.; Stanyer, L.; Yamamoto, M.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol. Open 2013, 2, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Wang, H.; Ding, Y.; Zhou, M.; Zhou, X.; Zhang, X.; Ding, K.; He, J.; Lu, X.; Xu, J.; et al. Genetic elimination of Nrf2 aggravates secondary complications except for vasospasm after experimental subarachnoid hemorrhage in mice. Brain Res. 2014, 1558, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Shih, A.Y.; Murphy, T.H.; Johnson, J.A. NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J. Biol. Chem. 2003, 278, 37948–37956. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Krishnan, J.; Ruckmani, K. Cigarette smoke and related risk factors in neurological disorders: An update. Biomed. Pharmacother. 2017, 85, 79–86. [Google Scholar]
- Santín-Márquez, R.; Alarcón-Aguilar, A.; López-Diazguerrero, N.E.; Chondrogianni, N.; Königsberg, M. Sulfoaphane—Role in aging and neurodegeneration. GeroScience 2019, 1–16. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Cuadrad, A. P 087—NRF2 controls proteostasis through the transcriptional regulation of autophagy. In Proceedings of the OCC World Congress and Annual SFRR-E Conference 2017 Metabolic Stress and Redox Regulation, Berlin, Germany, 21–23 June 2017; p. S47. [Google Scholar]
- Pajares, M.; Rojo, A.I.; Arias, E.; Diaz-Carretero, A.; Cuervo, A.M.; Cuadrado, A. Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 2018, 14, 1310–1322. [Google Scholar] [CrossRef]
- Niture, S.K.; Jaiswal, A.K. Nrf2 Protein Up-regulates Antiapoptotic Protein Bcl-2 and Prevents Cellular Apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef]
- Harada, N.; Kanayama, M.; Maruyama, A.; Yoshida, A.; Tazumi, K.; Hosoya, T.; Mimura, J.; Toki, T.; Maher, J.M.; Yamamoto, M. Nrf2 regulates ferroportin 1-mediated iron efflux and counteracts lipopolysaccharide-induced ferroportin 1 mRNA suppression in macrophages. Arch. Biochem. Biophys. 2011, 508, 101–109. [Google Scholar] [CrossRef]
- Sakata, H.; Niizuma, K.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Narasimhan, P.; Maier, C.M.; Nishiyama, Y.; Chan, P.H. Minocycline-preconditioned neural stem cells enhance neuroprotection after ischemic stroke in rats. J. Neurosci. 2012, 32, 3462–3473. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.-S.; Ueno, I.; Sakamoto, A.; Tong, K.I. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213. [Google Scholar] [CrossRef] [PubMed]
- Piantadosi, C.A.; Withers, C.M.; Bartz, R.R.; MacGarvey, N.C.; Fu, P.; Sweeney, T.E.; Welty-Wolf, K.E.; Suliman, H.B. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 2011, 286, 16374–16385. [Google Scholar] [CrossRef] [PubMed]
- Vomhof-DeKrey, E.E.; Picklo Sr, M.J. The Nrf2-antioxidant response element pathway: a target for regulating energy metabolism. J. Nutr. Biochem. 2012, 23, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, L.F.; Pedruzzi, L.M.; Stenvinkel, P.; Stockler-Pinto, M.B.; Daleprane, J.B.; Leite Jr, M.; Mafra, D. Nutritional strategies to modulate inflammation and oxidative stress pathways via activation of the master antioxidant switch Nrf2. Biochimie 2013, 95, 1525–1533. [Google Scholar] [CrossRef]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1–Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Cominacini, L.; Mozzini, C.; Garbin, U.; Pasini, A.; Stranieri, C.; Solani, E.; Vallerio, P.; Tinelli, I.A.; Pasini, A.F. Endoplasmic reticulum stress and Nrf2 signaling in cardiovascular diseases. Free Radic. Biol. Med. 2015, 88, 233–242. [Google Scholar] [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Plafker, K.S.; Nguyen, L.; Barneche, M.; Mirza, S.; Crawford, D.F.; Plafker, S.M. The ubiquitin conjugating enzyme, UbcM2, can regulate the stability and activity of the anti-oxidant transcription factor, Nrf2. J. Biol. Chem. 2010, 285, 23064–23074. [Google Scholar] [CrossRef]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2—An update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef]
- Xiang, M.; Namani, A.; Wu, S.; Wang, X. Nrf2: bane or blessing in cancer? J. Cancer Res. Clin. Oncol. 2014, 140, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: an update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Evrard-Todeschi, N.; Innamorato, N.G.; Cotte, A.; Jaworski, T.; Tobon-Velasco, J.C.; Devijver, H.; Garcia-Mayoral, M.F.; Van Leuven, F.; et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol. Cell Biol. 2012, 32, 3486–3499. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Chen, Y.; Hou, X.; Huang, M.; Jin, J. Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug Metab. Rev. 2016, 48, 541–567. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Cucullo, L. Pathobiology of tobacco smoking and neurovascular disorders: untied strings and alternative products. Fluids Barriers CNS 2015, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and-independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Strom, J.; Xu, B.; Tian, X.; Chen, Q.M. Nrf2 protects mitochondrial decay by oxidative stress. FASEB J. 2016, 30, 66–80. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Espada, S.; Rojo, A.I.; Salinas, M.; Cuadrado, A. The muscarinic M1 receptor activates Nrf2 through a signaling cascade that involves protein kinase C and inhibition of GSK-3beta: connecting neurotransmission with neuroprotection. J. Neurochem. 2009, 110, 1107–1119. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Palliyaguru, D.L.; Kensler, T.W. Frugal chemoprevention: targeting Nrf2 with foods rich in sulforaphane. Semin. Oncol. 2016, 43, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 system: An evolutionary journey through stressful space and time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef] [PubMed]
- Sajja, R.K.; Green, K.N.; Cucullo, L. Altered Nrf2 signaling mediates hypoglycemia-induced blood-brain barrier endothelial dysfunction in vitro. PLoS ONE 2015, 10, e0122358. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Jain, A.K.; Shelton, P.M.; Jaiswal, A.K. Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. J. Biol. Chem. 2017, 292, 2048. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Hirano, I.; Itoh, T.; Tanaka, M.; Miyajima, A.; Suzuki, A.; Motohashi, H.; Yamamoto, M. Nrf2 Enhances Cholangiocyte Expansion in Pten-Deficient Livers. Mol. Cell. Biol. 2014, 34, 900–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Waguri, S.; Sou, Y.-s.; Kageyama, S.; Hasegawa, J.; Ishimura, R.; Saito, T.; Yang, Y.; Kouno, T.; Fukutomi, T.; et al. Phosphorylation of p62 Activates the Keap1-Nrf2 Pathway during Selective Autophagy. Mol. Cell 2013, 51, 618–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Gregg, M.; Hashemi, F.; Schott, L.; Hughes, T.K. Corticotropin Releasing Factor (CRF) activation of NF-κB-directed transcription in leukocytes. Cell Mol. Neurobiol. 2006, 26, 1021–1036. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Rojo, A.I.; García-Yagüe, Á.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The Transcription Factor Nrf2 Is a Therapeutic Target against Brain Inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, L.-O.; Kipp, M.; Lucius, R.; Pufe, T.; Wruck, C.J. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflamm. Res. 2010, 59, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Lastres-Becker, I.; Cuadrado, A. Role of microglial redox balance in modulation of neuroinflammation. Curr. Opin. Neurol. 2009, 22, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Cuadrado, A.; Lastres-Becker, I.; Kirik, D.; Sahin, G.; Ulusoy, A.; Rábano, A. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet. 2012, 21, 3173–3192. [Google Scholar]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-Induced Expression of NAD(P)H:Quinone Oxidoreductase 1 and Heme Oxygenase-1 Protects against Excessive Inflammatory Responses in Human Monocytes. J. Immunol. 2008, 181, 6730–6737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 Talks, Who’s Listening? Antioxid. Redox Signal 2010, 13, 1649–1663. [Google Scholar] [CrossRef]
- Cuadrado, A.; Martín-Moldes, Z.; Ye, J.; Lastres-Becker, I. Transcription factors NRF2 and NF-κB are coordinated effectors of the Rho family, GTP-binding protein RAC1 during inflammation. J. Biol. Chem. 2014, 289, 15244–15258. [Google Scholar] [CrossRef]
- Sanlioglu, S.; Williams, C.M.; Samavati, L.; Butler, N.S.; Wang, G.; McCray, P.B.; Ritchie, T.C.; Hunninghake, G.W.; Zandi, E.; Engelhardt, J.F. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-α secretion through IKK regulation of NF-κB. J. Biol. Chem. 2001, 276, 30188–30198. [Google Scholar] [CrossRef]
- Zhang, H.; Davies, K.J.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohal, R.S.; Orr, W.C. The redox stress hypothesis of aging. Free Radic. Biol. Med. 2012, 52, 539–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngo, J.K.; Pomatto, L.C.; Davies, K.J. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013, 1, 258–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarantini, S.; Valcarcel-Ares, M.N.; Yabluchanskiy, A.; Tucsek, Z.; Hertelendy, P.; Kiss, T.; Gautam, T.; Zhang, X.A.; Sonntag, W.E.; de Cabo, R.; et al. Nrf2 Deficiency Exacerbates Obesity-Induced Oxidative Stress, Neurovascular Dysfunction, Blood-Brain Barrier Disruption, Neuroinflammation, Amyloidogenic Gene Expression, and Cognitive Decline in Mice, Mimicking the Aging Phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, H.; Davies, K.J.; Sioutas, C.; Finch, C.E.; Morgan, T.E.; Forman, H.J. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic. Biol. Med. 2012, 52, 2038–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Palacios, A.; Ostolga-Chavarria, M.; Zazueta, C.; Konigsberg, M. Nrf2: Molecular and epigenetic regulation during aging. Ageing Res. Rev. 2018, 47, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. In-vitro blood–brain barrier modeling: A review of modern and fast-advancing technologies. J. Cereb. Blood Flow Metab. 2018, 0271678X18788769. [Google Scholar] [CrossRef]
- Hasan, A.; Deeb, G.; Rahal, R.; Atwi, K.; Mondello, S.; Marei, H.E.; Gali, A.; Sleiman, E. Mesenchymal stem cells in the treatment of traumatic brain injury. Front. Neurol. 2017, 8, 28. [Google Scholar] [CrossRef]
- Semple, B.D.; Zamani, A.; Rayner, G.; Shultz, S.R.; Jones, N.C. Affective, neurocognitive and psychosocial disorders associated with traumatic brain injury and post-traumatic epilepsy. Neurobiol. Dis. 2019, 123, 27–41. [Google Scholar] [CrossRef]
- Dong, W.; Yang, B.; Wang, L.; Li, B.; Guo, X.; Zhang, M.; Jiang, Z.; Fu, J.; Pi, J.; Guan, D. Curcumin plays neuroprotective roles against traumatic brain injury partly via Nrf2 signaling. Toxicol. Appl. Pharmacol. 2018, 346, 28–36. [Google Scholar] [CrossRef]
- Angeloni, C.; Prata, C.; Vieceli Dalla Sega, F.; Piperno, R.; Hrelia, S. Traumatic brain injury and NADPH oxidase: a deep relationship. Oxidative Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Park, S.; Krause, J.S.; Banik, N.L. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem. Int. 2013, 62, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yan, H.; Ni, H.; Liang, W.; Jin, W. Expression of nuclear factor erythroid 2-related factor 2 following traumatic brain injury in the human brain. Neuroreport 2019, 30, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tian, M.; Wang, H.D.; Gao, C.C.; Zhu, L.; Lin, Y.X.; Fang, J.; Ding, K. Activation of the Nrf2-ARE signal pathway after blast induced traumatic brain injury in mice. Int. J. Neurosci. 2019, 129, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Suvanish Kumar, V.; Gopalakrishnan, A.; Naziroglu, M.; Rajanikant, G. Calcium ion–the key player in cerebral ischemia. Curr. Med. Chem. 2014, 21, 2065–2075. [Google Scholar] [CrossRef]
- Pradeep, H.; Diya, J.B.; Shashikumar, S.; Rajanikant, G.K. Oxidative stress–assassin behind the ischemic stroke. Folia Neuropathol. 2012, 50, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, D.; Yin, T.; Smalstig, E.B.; Hsu, M.A.; Panetta, J.; Little, S.; Clemens, J. Transcription factor nuclear factor-κB is activated in neurons after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2000, 20, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Kunz, A.; Abe, T.; Hochrainer, K.; Shimamura, M.; Anrather, J.; Racchumi, G.; Zhou, P.; Iadecola, C. Nuclear factor-κB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J. Neurosci. 2008, 28, 1649–1658. [Google Scholar] [CrossRef]
- Harari, O.A.; Liao, J.K. NF-κB and innate immunity in ischemic stroke. Ann. N Y Acad. Sci. 2010, 1207, 32–40. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, Y.; He, Q.; Li, L.; Xie, H.; Zhao, Y.; Zhao, J. Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav. Brain Res. 2018, 336, 32–39. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Villalba, H.; Prasad, S.; Liles, T.; Sifat, A.E.; Sajja, R.K.; Abbruscato, T.J.; Cucullo, L. Offsetting the impact of smoking and e-cigarette vaping on the cerebrovascular system and stroke injury: Is Metformin a viable countermeasure? Redox Biol. 2017, 13, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Locascio, L.M.; Dore, S. Critical Role of Nrf2 in Experimental Ischemic Stroke. Front. Pharm. 2019, 10, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.; Kaisar, M.A.; Cucullo, L. Unhealthy smokers: scopes for prophylactic intervention and clinical treatment. BMC Neurosci. 2017, 18, 70. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kang, J.; Li, H.; Su, J.; Wu, J.; Xu, Y.; Yu, H.; Xiang, X.; Yi, H.; Lu, Y. Regulation of endoplasmic reticulum stress in rat cortex by p62/ZIP through the Keap1-Nrf2-ARE signalling pathway after transient focal cerebral ischaemia. Brain Inj. 2013, 27, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, X.; Kim, Y.; Li, J.; Huang, S.; Saleem, S.; Li, R.C.; Xu, Y.; Dore, S.; Cao, W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic. Biol. Med. 2012, 52, 928–936. [Google Scholar] [CrossRef]
- Zhao, J.; Moore, A.N.; Redell, J.B.; Dash, P.K. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 2007, 27, 10240–10248. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Cui, L.; Wang, L.; Liu, H.; Ji, H.; Du, Y. Ursolic acid promotes the neuroprotection by activating Nrf2 pathway after cerebral ischemia in mice. Brain Res. 2013, 1497, 32–39. [Google Scholar] [CrossRef]
- Wang, H.; Zhong, D.; Chen, H.; Jin, J.; Liu, Q.; Li, G. NLRP3 inflammasome activates interleukin-23/interleukin-17 axis during ischaemia-reperfusion injury in cerebral ischaemia in mice. Life Sci. 2019, 227, 101–113. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Sivandzade, F.; Bhalerao, A.; Cucullo, L. Conventional and electronic cigarettes dysregulate the expression of iron transporters and detoxifying enzymes at the brain vascular endothelium: In vivo evidence of a gender-specific cellular response to chronic cigarette smoke exposure. Neurosci. Lett. 2018, 682, 1–9. [Google Scholar] [CrossRef]
- Carvalho, C.; Machado, N.; Mota, P.C.; Correia, S.C.; Cardoso, S.; Santos, R.X.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Type 2 diabetic and Alzheimer’s disease mice present similar behavioral, cognitive, and vascular anomalies. J. Alzheimer’s Dis. 2013, 35, 623–635. [Google Scholar] [CrossRef]
- Wevers, N.R.; de Vries, H.E. Morphogens and blood-brain barrier function in health and disease. Tissue Barriers 2016, 4, e1090524. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Et Biophys. Acta (BBA)-Mol. Basis. Dis. 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Haapasalo, A.; Hiltunen, M.; Soininen, H.; Alafuzoff, I. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2012, 96, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood–brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1822, 822–829. [Google Scholar] [CrossRef]
- Bae, E.-J.; Ho, D.-H.; Park, E.; Jung, J.W.; Cho, K.; Hong, J.H.; Lee, H.-J.; Kim, K.P.; Lee, S.-J. Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of α-synuclein. Antioxid. Redox Signal 2013, 18, 770–783. [Google Scholar] [CrossRef]
- Li, X.-H.; Li, C.-Y.; Lu, J.-M.; Tian, R.-B.; Wei, J. Allicin ameliorates cognitive deficits ageing-induced learning and memory deficits through enhancing of Nrf2 antioxidant signaling pathways. Neurosci. Lett. 2012, 514, 46–50. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Maghsoudi, N.; Khodagholi, F. Stabilization of transcription factor Nrf2 by tBHQ prevents oxidative stress-induced amyloid β formation in NT2N neurons. Biochimie 2010, 92, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Garcia-Yague, A.J.; Buendia, I.; Van Leuven, F.; Yamamoto, M.; Lopez, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Manczak, M.; Calkins, M.J.; Truong, Q.; Reddy, T.P.; Reddy, A.P.; Shirendeb, U.; Lo, H.-H.; Rabinovitch, P.S.; Reddy, P.H. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 2012, 21, 2973–2990. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Pajares, M.; Rada, P.; Nunez, A.; Nevado-Holgado, A.J.; Killik, R.; Van Leuven, F.; Ribe, E.; Lovestone, S.; Yamamoto, M.; et al. NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology. Redox Biol. 2017, 13, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Näsström, T.; Fagerqvist, T.; Barbu, M.; Karlsson, M.; Nikolajeff, F.; Kasrayan, A.; Ekberg, M.; Lannfelt, L.; Ingelsson, M.; Bergström, J. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of α-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic. Biol. Med. 2011, 50, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid Redox Signal 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.Y.; Kim, J.H.; Moon, M.K.; Han, S.-H.; Yeon, S.K.; Choi, J.W.; Jang, B.K.; Song, H.J.; Kang, Y.G.; Kim, J.W. Discovery of vinyl sulfones as a novel class of neuroprotective agents toward Parkinson’s disease therapy. J. Med. Chem. 2014, 57, 1473–1487. [Google Scholar] [CrossRef]
- Gan, L.; Vargas, M.R.; Johnson, D.A.; Johnson, J.A. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J. Neurosci. 2012, 32, 17775–17787. [Google Scholar] [CrossRef]
- He, Q.; Song, N.; Jia, F.; Xu, H.; Yu, X.; Xie, J.; Jiang, H. Role of α-synuclein aggregation and the nuclear factor E2-related factor 2/heme oxygenase-1 pathway in iron-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2013, 45, 1019–1030. [Google Scholar] [CrossRef]
- Barone, M.C.; Sykiotis, G.P.; Bohmann, D. Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Models Mech. 2011, 4, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.N.; Yanxun, V.Y.; Gundemir, S.; Jo, C.; Cui, M.; Tieu, K.; Johnson, G.V. Impaired mitochondrial dynamics and Nrf2 signaling contribute to compromised responses to oxidative stress in striatal cells expressing full-length mutant huntingtin. PLoS ONE 2013, 8, e57932. [Google Scholar] [CrossRef] [PubMed]
- Stack, C.; Ho, D.; Wille, E.; Calingasan, N.Y.; Williams, C.; Liby, K.; Sporn, M.; Dumont, M.; Beal, M.F. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic. Biol. Med. 2010, 49, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Hands, S.; Sajjad, M.U.; Newton, M.J.; Wyttenbach, A. In vitro and in vivo aggregation of a fragment of huntingtin protein directly causes free radical production. J. Biol. Chem. 2011, 286, 44512–44520. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, A.S.; Arrasate, M.; Barmada, S.; Ando, D.M.; Sharma, P.; Shaby, B.A.; Finkbeiner, S. Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat. Chem. Biol. 2013, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Tanaka, K.; Yanagisawa, Y.; Yasutake, K.; Hadano, S.; Yoshii, F.; Hirayama, N.; Ikeda, J.-E. A novel small molecule, N-(4-(2-pyridyl)(1,3-thiazol-2-yl))-2-(2,4,6-trimethylphenoxy) acetamide, selectively protects against oxidative stress-induced cell death by activating the Nrf2–ARE pathway: Therapeutic implications for ALS. Free Radic. Biol. Med. 2012, 53, 2028–2042. [Google Scholar] [CrossRef] [PubMed]
- Mead, R.J.; Higginbottom, A.; Allen, S.P.; Kirby, J.; Bennett, E.; Barber, S.C.; Heath, P.R.; Coluccia, A.; Patel, N.; Gardner, I. S [+] Apomorphine is a CNS penetrating activator of the Nrf2-ARE pathway with activity in mouse and patient fibroblast models of amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2013, 61, 438–452. [Google Scholar] [CrossRef]
- Liebner, S.; Czupalla, C.J.; Wolburg, H. Current concepts of blood-brain barrier development. Int. J. Dev. Biol. 2011, 55, 467–476. [Google Scholar] [CrossRef]
- Alfieri, A.; Srivastava, S.; Siow, R.C.; Modo, M.; Fraser, P.A.; Mann, G.E. Targeting the Nrf2–Keap1 antioxidant defence pathway for neurovascular protection in stroke. J. Physiol. 2011, 589, 4125–4136. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef]
- Naik, P.; Sajja, R.K.; Prasad, S.; Cucullo, L. Effect of full flavor and denicotinized cigarettes exposure on the brain microvascular endothelium: a microarray-based gene expression study using a human immortalized BBB endothelial cell line. BMC Neurosci. 2015, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-Y.; Wang, H.-D.; Xu, J.-G.; Ding, K.; Li, T. Deletion of Nrf2 exacerbates oxidative stress after traumatic brain injury in mice. Cell. Mol. Neurobiol. 2015, 35, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Campos, C.R.; Peart, J.C.; Smith, L.K.; Boni, J.L.; Cannon, R.E.; Miller, D.S. Nrf2 upregulates ATP binding cassette transporter expression and activity at the blood–brain and blood–spinal cord barriers. J. Neurosci. 2014, 34, 8585–8593. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Fang, Q.; Zhang, J.; Zhou, D.; Wang, Z. Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J. Neurosci. Res. 2011, 89, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Petri, S.; Körner, S.; Kiaei, M. Nrf2/ARE signaling pathway: key mediator in oxidative stress and potential therapeutic target in ALS. Neurol. Res. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int. J. Mol. Sci. 2012, 13, 11783–11803. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Sajja, R.K.; Naik, P.; Cucullo, L. Diabetes mellitus and blood-brain barrier dysfunction: an overview. J. Pharmacovigil. 2014, 2, 125. [Google Scholar] [PubMed]
- Wang, W.; Wu, Y.; Zhang, G.; Fang, H.; Wang, H.; Zang, H.; Xie, T.; Wang, W. Activation of Nrf2-ARE signal pathway protects the brain from damage induced by epileptic seizure. Brain Res. 2014, 1544, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, A.; Srivastava, S.; Siow, R.C.; Cash, D.; Modo, M.; Duchen, M.R.; Fraser, P.A.; Williams, S.C.; Mann, G.E. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic. Biol. Med. 2013, 65, 1012–1022. [Google Scholar] [CrossRef]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharm. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharm. Exp. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Yaribeygi, H.; Panahi, Y.; Javadi, B.; Sahebkar, A. The Underlying Role of Oxidative Stress in Neurodegeneration: A Mechanistic Review. CNS Neurol. Disord. Drug Targets 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Mao, X.; Liu, A.; Gao, X.; Chen, X.; Ye, M.; Ye, J.; Liu, P.; Xu, S.; Liu, J. Osthole, a natural coumarin improves cognitive impairments and BBB dysfunction after transient global brain ischemia in C57 BL/6J mice: involvement of Nrf2 pathway. Neurochem. Res. 2015, 40, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takagi, T.; Kitashoji, A.; Yamauchi, K.; Shimazawa, M.; Hara, H. Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol. Dis. 2016, 89, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Suwanwela, N.C.; Patumraj, S. Curcumin by down-regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc. Res. 2016, 106, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a potential protective phytochemical against neurodegenerative diseases. Oxidative Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Yang, T.; Li, X.; Lei, X.; Sun, Y.; Zhao, Y.; Zhang, W.; Gao, Y.; Sun, B.; Zhang, F. Protective effects of sulforaphane in experimental vascular cognitive impairment: Contribution of the Nrf2 pathway. J. Cereb. Blood Flow Metab. 2018, 0271678X18764083. [Google Scholar] [CrossRef] [PubMed]
- Soane, L.; Li Dai, W.; Fiskum, G.; Bambrick, L.L. Sulforaphane protects immature hippocampal neurons against death caused by exposure to hemin or to oxygen and glucose deprivation. J. Neurosci. Res. 2010, 88, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; He, Q.; Zheng, J.; Li, L.Y.; Hou, Y.H.; Song, F.Z. Sulforaphane improves outcomes and slows cerebral ischemic/reperfusion injury via inhibition of NLRP3 inflammasome activation in rats. Int. Immunopharmacol. 2017, 45, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.M.; Gillespie, S.; Becker, F.; Vital, S.A.; Nguyen, V.; Alexander, J.S.; Evans, P.C.; Gavins, F.N. Sulforaphane induces neurovascular protection against a systemic inflammatory challenge via both Nrf2-dependent and independent pathways. Vasc. Pharmacol. 2016, 85, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V. Glucosinolates and isothiocyanates in health and disease. Trends Mol. Med. 2012, 18, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wen, L.; Dong, M.; Lu, X. Sulforaphane activates the cerebral vascular Nrf2–ARE pathway and suppresses inflammation to attenuate cerebral vasospasm in rat with subarachnoid hemorrhage. Brain Res. 2016, 1653, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Takaya, K.; Suzuki, T.; Motohashi, H.; Onodera, K.; Satomi, S.; Kensler, T.W.; Yamamoto, M. Validation of the multiple sensor mechanism of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2012, 53, 817–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Noel, J.; Kaplan, H.J.; Dean, D.C. Circulating reactive oxidant causes apoptosis of retinal pigment epithelium and cone photoreceptors in the mouse central retina. Ophthalmol. Eye Dis. 2011, 3, 45–54. [Google Scholar] [CrossRef]
- Muller, T.; Hengstermann, A. Nrf2: friend and foe in preventing cigarette smoking-dependent lung disease. Chem. Res. Toxicol. 2012, 25, 1805–1824. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; He, X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharm. Rev. 2012, 64, 1055–1081. [Google Scholar] [CrossRef]
- Kaisar, M.A.; Prasad, S.; Cucullo, L. Protecting the BBB endothelium against cigarette smoke-induced oxidative stress using popular antioxidants: Are they really beneficial? Brain Res. 2015, 1627, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-Y.; Kuan, Y.-H.; Li, J.-R.; Chen, W.-Y.; Ou, Y.-C.; Pan, H.-C.; Liao, S.-L.; Raung, S.-L.; Chang, C.-J.; Chen, C.-J. Docosahexaenoic acid reduces cellular inflammatory response following permanent focal cerebral ischemia in rats. J. Nutr. Biochem. 2013, 24, 2127–2137. [Google Scholar] [CrossRef]
- Ren, J.; Fan, C.; Chen, N.; Huang, J.; Yang, Q. Resveratrol pretreatment attenuates cerebral ischemic injury by upregulating expression of transcription factor Nrf2 and HO-1 in rats. Neurochem. Res. 2011, 36, 2352. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, B.; Zhang, X.; Zhang, L.; Bai, X.; Zhao, X.; Chen, L.; Cui, L.; Zhu, C.; Wang, L. Bicyclol upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res. Bull. 2014, 100, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M. Cellular response to cigarette smoke and oxidants: adapting to survive. Proc. Am. Thorac. Soc. 2010, 7, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Petrache, I. Pathogenesis of chronic obstructive pulmonary disease. J. Clin. Investig. 2012, 122, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. Assessing the protective effect of rosiglitazone against electronic cigarette/tobacco smoke-induced blood–brain barrier impairment. BMC Neurosci. 2019, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.; Li, W.; Abdul, Y.; Jackson, L.; Dong, G.; Jamil, S.; Filosa, J.; Fagan, S.C.; Ergul, A. NLRP3 inflammasome inhibition with MCC950 improves diabetes-mediated cognitive impairment and vasoneuronal remodeling after ischemia. Pharm. Res. 2019, 142, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ma, C.; Meng, C.J.; Zhu, G.Q.; Sun, X.B.; Huo, L.; Zhang, J.; Liu, H.X.; He, W.C.; Shen, X.M. Melatonin activates the Nrf2-ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J. Pineal Res. 2012, 53, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, G.; Li, Y.; Wang, Y.; Chen, X.; Gu, X.; Zhang, Z.; Wang, Y.; Yang, G.Y. Metformin attenuates blood-brain barrier disruption in mice following middle cerebral artery occlusion. J. Neuroinflammation 2014, 11, 177. [Google Scholar] [CrossRef]
- Jin, Q.; Cheng, J.; Liu, Y.; Wu, J.; Wang, X.; Wei, S.; Zhou, X.; Qin, Z.; Jia, J.; Zhen, X. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav. Immun. 2014, 40, 131–142. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Tanokashira, D.; Kurata, E.; Fukuokaya, W.; Kawabe, K.; Kashiwada, M.; Takeuchi, H.; Nakazato, M.; Taguchi, A. Metformin treatment ameliorates diabetes-associated decline in hippocampal neurogenesis and memory via phosphorylation of insulin receptor substrate 1. FEBS Open Biol. 2018, 8, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Pryor, R.; Cabreiro, F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem. J. 2015, 471, 307–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ming, M.; Sinnett-Smith, J.; Wang, J.; Soares, H.P.; Young, S.H.; Eibl, G.; Rozengurt, E. Dose-dependent AMPK-dependent and independent mechanisms of berberine and metformin inhibition of mTORC1, ERK, DNA synthesis and proliferation in pancreatic cancer cells. PLoS ONE 2014, 9, e114573. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Malinowski, J.M.; Bolesta, S. Rosiglitazone in the treatment of type 2 diabetes mellitus: A critical review. Clin 2000, 22, 1151–1168, discussion 1149–1150. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD (P) H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Z.; Liu, J.Z.; Hu, J.X.; Chen, H.L.; Li, W.L.; Hai, C.X. Double antioxidant activities of rosiglitazone against high glucose-induced oxidative stress in hepatocyte. Toxicol. Vitr. 2011, 25, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Park, J.H.; Jang, S.J.; Koh, H.C. Rosiglitazone inhibits chlorpyrifos-induced apoptosis via modulation of the oxidative stress and inflammatory response in SH-SY5Y cells. Toxicol. Appl. Pharmacol. 2014, 278, 159–171. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin–a future therapy for neurodegenerative diseases. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef]
- Sayan-Ozacmak, H.; Ozacmak, V.H.; Barut, F.; Jakubowska-Dogru, E. Rosiglitazone treatment reduces hippocampal neuronal damage possibly through alleviating oxidative stress in chronic cerebral hypoperfusion. Neurochem. Int. 2012, 61, 287–290. [Google Scholar] [CrossRef]
- Kadam, L.; Gomez-Lopez, N.; Mial, T.N.; Kohan-Ghadr, H.R.; Drewlo, S. Rosiglitazone Regulates TLR4 and Rescues HO-1 and NRF2 Expression in Myometrial and Decidual Macrophages in Inflammation-Induced Preterm Birth. Reprod. Sci. 2017, 24, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Seldon, M.P.; Gregoire, I.P.; Vassilevskaia, T.; Berberat, P.O.; Yu, J.; Tsui, T.Y.; Bach, F.H. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J. Immunol. 2004, 172, 3553–3563. [Google Scholar] [CrossRef] [PubMed]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivandzade, F.; Bhalerao, A.; Cucullo, L. Cerebrovascular and Neurological Disorders: Protective Role of NRF2. Int. J. Mol. Sci. 2019, 20, 3433. https://doi.org/10.3390/ijms20143433
Sivandzade F, Bhalerao A, Cucullo L. Cerebrovascular and Neurological Disorders: Protective Role of NRF2. International Journal of Molecular Sciences. 2019; 20(14):3433. https://doi.org/10.3390/ijms20143433
Chicago/Turabian StyleSivandzade, Farzane, Aditya Bhalerao, and Luca Cucullo. 2019. "Cerebrovascular and Neurological Disorders: Protective Role of NRF2" International Journal of Molecular Sciences 20, no. 14: 3433. https://doi.org/10.3390/ijms20143433
APA StyleSivandzade, F., Bhalerao, A., & Cucullo, L. (2019). Cerebrovascular and Neurological Disorders: Protective Role of NRF2. International Journal of Molecular Sciences, 20(14), 3433. https://doi.org/10.3390/ijms20143433