Glucagon, GLP-1 and Thermogenesis

 ,
, 
 and
and 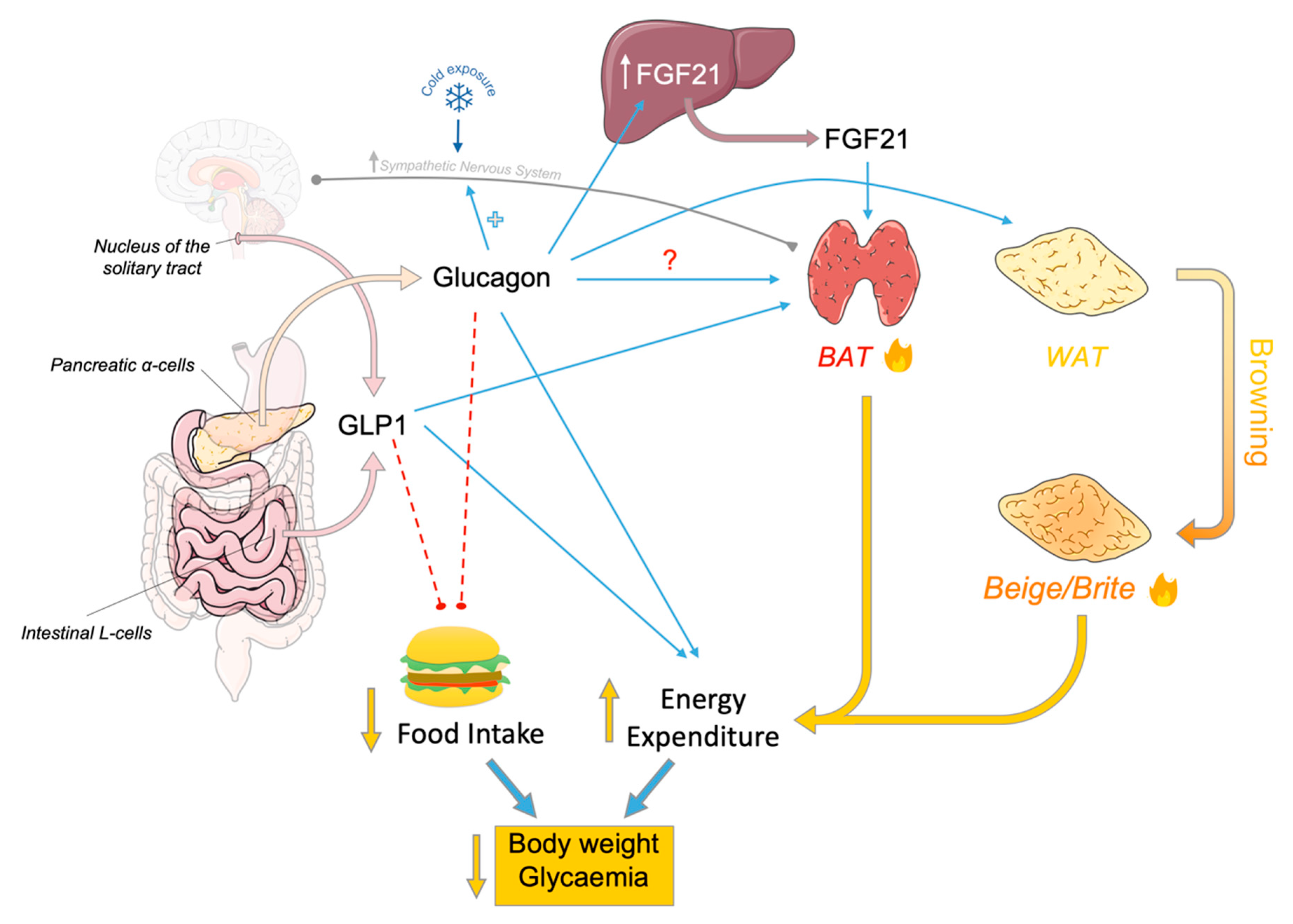{kind=link}
Abstract
:1. Introduction
2. Glucagon and Thermogenesis
3. Effects of GLP-1 on the Energy Balance
Central Effects of GLP-1
4. New Therapeutic Approaches of Glucagon and GLP-1 Against Obesity
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Gomis, R.; Claret, M. Hypothalamic and brainstem neuronal circuits controlling homeostatic energy balance. J. Endocrinol. 2014, 220, T25–T46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; et al. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar] [CrossRef]
- Contreras, C.; Gonzalez, F.; Ferno, J.; Dieguez, C.; Rahmouni, K.; Nogueiras, R.; Lopez, M. The brain and brown fat. Ann. Med. 2015, 47, 150–168. [Google Scholar] [CrossRef]
- Brakenhielm, E.; Cao, R.; Gao, B.; Angelin, B.; Cannon, B.; Parini, P.; Cao, Y. Angiogenesis inhibitor, tnp-470, prevents diet-induced and genetic obesity in mice. Circ. Res. 2004, 94, 1579–1588. [Google Scholar] [CrossRef]
- Morrison, S.F.; Nakamura, K. Central neural pathways for thermoregulation. Front. Biosci.(Landmark.Ed.) 2011, 16, 74–104. [Google Scholar] [CrossRef] [Green Version]
- Labbe, S.M.; Caron, A.; Lanfray, D.; Monge-Rofarello, B.; Bartness, T.J.; Richard, D. Hypothalamic control of brown adipose tissue thermogenesis. Front. Syst. Neurosci. 2015, 9, 150. [Google Scholar] [CrossRef]
- Contreras, C.; Gonzalez-Garcia, I.; Seoane-Collazo, P.; Martinez-Sanchez, N.; Linares-Pose, L.; Rial-Pensado, E.; Ferno, J.; Tena-Sempere, M.; Casals, N.; Dieguez, C.; et al. Reduction of hypothalamic endoplasmic reticulum stress activates browning of white fat and ameliorates obesity. Diabetes 2017, 66, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.A.; D’Alessio, D.A. Physiology of proglucagon peptides: Role of glucagon and GLP-1 in health and disease. Physiol. Rev. 2015, 95, 513–548. [Google Scholar] [CrossRef] [PubMed]
- Campos, R.V.; Lee, Y.C.; Drucker, D.J. Divergent tissue-specific and developmental expression of receptors for glucagon and glucagon-like peptide-1 in the mouse. Endocrinology 1994, 134, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, M.; Tastenoy, M.; Vertongen, P.; Robberecht, P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol. Cell Endocrinol. 1994, 105, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J. The biology of incretin hormones. Cell Metab. 2006, 3, 153–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stunkard, A.J.; Van Itallie, T.B.; Reis, B.B. The mechanism of satiety: Effect of glucagon on gastric hunger contractions in man. Proc. Soc. Exp. Biol. Med. 1955, 89, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Schulman, J.L.; Carleton, J.L.; Whitney, G.; Whitehorn, J.C. Effect of glucagon on food intake and body weight in man. J. Appl. Physiol. 1957, 11, 419–421. [Google Scholar] [CrossRef]
- Penick, S.B.; Hinkle, L.E., Jr. Depression of food intake induced in healthy subjects by glucagon. N. Engl. J. Med. 1961, 264, 893–897. [Google Scholar] [CrossRef]
- Quinones, M.; Al-Massadi, O.; Gallego, R.; Ferno, J.; Dieguez, C.; Lopez, M.; Nogueiras, R. Hypothalamic camkkbeta mediates glucagon anorectic effect and its diet-induced resistance. Mol. Metab. 2015, 4, 961–970. [Google Scholar] [CrossRef]
- Geary, N.; Smith, G.P. Pancreatic glucagon fails to inhibit sham feeding in the rat. Peptides 1982, 3, 163–166. [Google Scholar] [CrossRef]
- Geary, N.; Le Sauter, J.; Noh, U. Glucagon acts in the liver to control spontaneous meal size in rats. Am. J. Physiol. 1993, 264, R116–R122. [Google Scholar] [CrossRef] [PubMed]
- Davidson, I.W.F.; Salter, J.M.; Best, C.H. The effect of glucagon on the metabolic rate of rats. Am. J. Clin. Nutr. 1960, 8, 540–546. [Google Scholar] [CrossRef]
- SALTER, J.M. Metabolic effects of glucagon in the wistar rat. Am. J. Clin. Nutr. 1960, 8, 535–539. [Google Scholar] [CrossRef]
- Chan, E.K.; Mackey, M.A.; Snover, D.C.; Schneider, P.D.; Rucker, R.D., Jr.; Allen, C.E.; Buchwald, H. Suppression of weight gain by glucagon in obese zucker rats. Exp. Mol. Pathol. 1984, 40, 320–327. [Google Scholar] [CrossRef]
- Nair, K.S. Hyperglucagonemia increases resting metabolic rate in man during insulin deficiency. J. Clin. Endocrinol. Metab. 1987, 64, 896–901. [Google Scholar] [CrossRef] [PubMed]
- Joel, C.D. Stimulation of metabolism of rat brown adipose tissue by addition of lipolytic hormones in vitro. J. Biol. Chem. 1966, 241, 814–821. [Google Scholar] [PubMed]
- Kuroshima, A.; Yahata, T. Thermogenic responses of brown adipocytes to noradrenaline and glucagon in heat-acclimated and cold-acclimated rats. Jpn. J. Physiol. 1979, 29, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Doi, K.; Kuroshima, A. Modified metabolic responsiveness to glucagon in cold-acclimated and heat-acclimated rats. Life Sci. 1982, 30, 785–791. [Google Scholar] [CrossRef]
- Seitz, H.J.; Krone, W.; Wilke, H.; Tarnowski, W. Rapid rise in plasma glucagon induced by acute cold exposure in man and rat. Pflugers Arch. 1981, 389, 115–120. [Google Scholar] [CrossRef]
- Helman, A.; Gilbert, M.; Pfister-Lemaire, N.; Reach, G.; Assan, R. Glucagon and insulin secretion and their biological activities in hypothermic rats. Endocrinology 1984, 115, 1722–1728. [Google Scholar] [CrossRef]
- Edwards, C.I.; Howland, R.J. Adaptive changes in insulin and glucagon secretion during cold acclimation in the rat. Am. J. Physiol. 1986, 250, E669–E676. [Google Scholar] [CrossRef] [PubMed]
- Habara, Y.; Kuroshima, A. Changes in glucagon and insulin contents of brown adipose tissue after temperature acclimation in rats. Jpn J. Physiol. 1983, 33, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.E.; DiMarchi, R.D.; Tschop, M.H.; Finan, B.; Campbell, J.E. Targeting the incretin/glucagon system with triagonists to treat diabetes. Endocr. Rev. 2018, 39, 719–738. [Google Scholar] [CrossRef] [PubMed]
- Tschop, M.H.; Finan, B.; Clemmensen, C.; Gelfanov, V.; Perez-Tilve, D.; Muller, T.D.; DiMarchi, R.D. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 2016, 24, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Ottaway, N.; Patterson, J.T.; Gelfanov, V.; Smiley, D.; Gidda, J.; Findeisen, H.; Bruemmer, D.; Drucker, D.J.; Chaudhary, N.; et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat. Chem. Biol. 2009, 5, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Day, J.W.; Gelfanov, V.; Smiley, D.; Carrington, P.E.; Eiermann, G.; Chicchi, G.; Erion, M.D.; Gidda, J.; Thornberry, N.A.; Tschop, M.H.; et al. Optimization of co-agonism at GLP-1 and glucagon receptors to safely maximize weight reduction in dio-rodents. Biopolymers 2012, 98, 443–450. [Google Scholar] [CrossRef]
- Clemmensen, C.; Chabenne, J.; Finan, B.; Sullivan, L.; Fischer, K.; Kuchler, D.; Sehrer, L.; Ograjsek, T.; Hofmann, S.M.; Schriever, S.C.; et al. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes 2014, 63, 1422–1427. [Google Scholar] [CrossRef]
- Kinoshita, K.; Ozaki, N.; Takagi, Y.; Murata, Y.; Oshida, Y.; Hayashi, Y. Glucagon is essential for adaptive thermogenesis in brown adipose tissue. Endocrinology 2014, 155, 3484–3492. [Google Scholar] [CrossRef]
- Beaudry, J.L.; Kaur, K.D.; Varin, E.M.; Baggio, L.L.; Cao, X.; Mulvihill, E.E.; Stern, J.H.; Campbell, J.E.; Scherer, P.E.; Drucker, D.J. The brown adipose tissue glucagon receptor is functional but not essential for control of energy homeostasis in mice. Mol. Metab. 2019, 22, 37–48. [Google Scholar] [CrossRef]
- Dicker, A.; Zhao, J.; Cannon, B.; Nedergaard, J. Apparent thermogenic effect of injected glucagon is not due to a direct effect on brown fat cells. Am. J. Physiol. 1998, 275, R1674–R1682. [Google Scholar] [CrossRef]
- Habegger, K.M.; Stemmer, K.; Cheng, C.; Muller, T.D.; Heppner, K.M.; Ottaway, N.; Holland, J.; Hembree, J.L.; Smiley, D.; Gelfanov, V.; et al. Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes 2013, 62, 1453–1463. [Google Scholar] [CrossRef] [PubMed]
- Giralt, M.; Gavalda-Navarro, A.; Villarroya, F. Fibroblast growth factor-21, energy balance and obesity. Mol. Cell. Endocrinol. 2015, 418 Pt 1, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Nason, S.; Holleman, C.; Pepin, M.; Wilson, L.; Berryhill, T.F.; Wende, A.R.; Steele, C.; Young, M.E.; Barnes, S.; et al. Glucagon receptor signaling regulates energy metabolism via hepatic farnesoid x receptor and fibroblast growth factor 21. Diabetes 2018, 67, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Calles-Escandon, J. Insulin dissociates hepatic glucose cycling and glucagon-induced thermogenesis in man. Metabolism 1994, 43, 1000–1005. [Google Scholar] [CrossRef]
- Salem, V.; Izzi-Engbeaya, C.; Coello, C.; Thomas, D.B.; Chambers, E.S.; Comninos, A.N.; Buckley, A.; Win, Z.; Al-Nahhas, A.; Rabiner, E.A.; et al. Glucagon increases energy expenditure independently of brown adipose tissue activation in humans. Diabetes Obes. Metab. 2016, 18, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and beige fat: Physiological roles beyond heat generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef]
- Townsend, L.K.; Medak, K.D.; Knuth, C.M.; Peppler, W.T.; Charron, M.J.; Wright, D.C. Loss of glucagon signaling alters white adipose tissue browning. FASEB J. 2019, 33, 4824–4835. [Google Scholar] [CrossRef]
- Mojsov, S.; Heinrich, G.; Wilson, I.B.; Ravazzola, M.; Orci, L.; Habener, J.F. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J. Biol. Chem. 1986, 261, 11880–11889. [Google Scholar]
- Holst, J.J. The physiology of glucagon-like peptide 1. Physiol. Rev. 2007, 87, 1409–1439. [Google Scholar] [CrossRef]
- Cork, S.C.; Richards, J.E.; Holt, M.K.; Gribble, F.M.; Reimann, F.; Trapp, S. Distribution and characterisation of glucagon-like peptide-1 receptor expressing cells in the mouse brain. Mol. Metab. 2015, 4, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L. Minireview: Finding the sweet spot: Peripheral versus central glucagon-like peptide 1 action in feeding and glucose homeostasis. Endocrinology 2009, 150, 2997–3001. [Google Scholar] [CrossRef] [PubMed]
- Kakei, M.; Yada, T.; Nakagawa, A.; Nakabayashi, H. Glucagon-like peptide-1 evokes action potentials and increases cytosolic ca2+ in rat nodose ganglion neurons. Auton. Neurosci. 2002, 102, 39–44. [Google Scholar] [CrossRef]
- Nakagawa, A.; Satake, H.; Nakabayashi, H.; Nishizawa, M.; Furuya, K.; Nakano, S.; Kigoshi, T.; Nakayama, K.; Uchida, K. Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton. Neurosci. 2004, 110, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Sisley, S.; Gutierrez-Aguilar, R.; Scott, M.; D’Alessio, D.A.; Sandoval, D.A.; Seeley, R.J. Neuronal glp1r mediates liraglutide’s anorectic but not glucose-lowering effect. J. Clin. Investig. 2014, 124, 2456–2463. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.A.; Ayala, J.E.; Smouse, H.; Landivar-Rocha, A.; Brown, J.D.; Drucker, D.J.; Stoffers, D.A.; Sandoval, D.A.; Seeley, R.J.; Ayala, J.E. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes 2017, 66, 372–384. [Google Scholar] [CrossRef]
- Manning, S.; Pucci, A.; Batterham, R.L. GLP-1: A mediator of the beneficial metabolic effects of bariatric surgery? Physiology (Bethesda) 2015, 30, 50–62. [Google Scholar] [CrossRef]
- Trapp, S.; Cork, S.C. Ppg neurons of the lower brain stem and their role in brain GLP-1 receptor activation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R795–R804. [Google Scholar] [CrossRef]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, W.L.; Habener, J.F. Glucagon-like peptide i stimulates insulin gene expression and increases cyclic amp levels in a rat islet cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar] [CrossRef]
- Kreymann, B.; Williams, G.; Ghatei, M.A.; Bloom, S.R. Glucagon-like peptide-1 7-36: A physiological incretin in man. Lancet 1987, 2, 1300–1304. [Google Scholar] [CrossRef]
- Mojsov, S.; Weir, G.C.; Habener, J.F. Insulinotropin: Glucagon-like peptide i (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J. Clin. Investig. 1987, 79, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Alcantara, A.I.; Morales, M.; Delgado, E.; Lopez-Delgado, M.I.; Clemente, F.; Luque, M.A.; Malaisse, W.J.; Valverde, I.; Villanueva-Penacarrillo, M.L. Exendin-4 agonist and exendin(9-39)amide antagonist of the GLP-1(7-36)amide effects in liver and muscle. Arch. Biochem. Biophys. 1997, 341, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Valverde, I.; Morales, M.; Clemente, F.; Lopez-Delgado, M.I.; Delgado, E.; Perea, A.; Villanueva-Penacarrillo, M.L. Glucagon-like peptide 1: A potent glycogenic hormone. FEBS Lett. 1994, 349, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Prigeon, R.L.; Quddusi, S.; Paty, B.; D’Alessio, D.A. Suppression of glucose production by GLP-1 independent of islet hormones: A novel extrapancreatic effect. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E701–E707. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Shin, S.; Shigihara, T.; Hahm, E.; Liu, M.J.; Han, J.; Yoon, J.W.; Jun, H.S. Glucagon-like peptide-1 gene therapy in obese diabetic mice results in long-term cure of diabetes by improving insulin sensitivity and reducing hepatic gluconeogenesis. Diabetes 2007, 56, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Idris, I.; Patiag, D.; Gray, S.; Donnelly, R. Exendin-4 increases insulin sensitivity via a pi-3-kinase-dependent mechanism: Contrasting effects of GLP-1. Biochem. Pharmacol. 2002, 63, 993–996. [Google Scholar] [CrossRef]
- Gonzalez, N.; Acitores, A.; Sancho, V.; Valverde, I.; Villanueva-Penacarrillo, M.L. Effect of GLP-1 on glucose transport and its cell signalling in human myocytes. Regul. Pept. 2005, 126, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Willms, B.; Werner, J.; Holst, J.J.; Orskov, C.; Creutzfeldt, W.; Nauck, M.A. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: Effects of exogenous glucagon-like peptide-1 (GLP-1)-(7-36) amide in type 2 (noninsulin-dependent) diabetic patients. J. Clin. Endocrinol. Metab. 1996, 81, 327–332. [Google Scholar] [PubMed]
- Flint, A.; Raben, A.; Astrup, A.; Holst, J.J. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J. Clin. Investig. 1998, 101, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Gutzwiller, J.P.; Goke, B.; Drewe, J.; Hildebrand, P.; Ketterer, S.; Handschin, D.; Winterhalder, R.; Conen, D.; Beglinger, C. Glucagon-like peptide-1: A potent regulator of food intake in humans. Gut 1999, 44, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Rodriquez de Fonseca, F.; Navarro, M.; Alvarez, E.; Roncero, I.; Chowen, J.A.; Maestre, O.; Gomez, R.; Munoz, R.M.; Eng, J.; Blazquez, E. Peripheral versus central effects of glucagon-like peptide-1 receptor agonists on satiety and body weight loss in zucker obese rats. Metabolism 2000, 49, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Ronveaux, C.C.; Tome, D.; Raybould, H.E. Glucagon-like peptide 1 interacts with ghrelin and leptin to regulate glucose metabolism and food intake through vagal afferent neuron signaling. J. Nutr. 2015, 145, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L.; Baskin, D.G.; Schwartz, M.W. Evidence that intestinal glucagon-like peptide-1 plays a physiological role in satiety. Endocrinology 2009, 150, 1680–1687. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L.; Baskin, D.G.; Schwartz, M.W. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes 2006, 55, 3387–3393. [Google Scholar] [CrossRef] [PubMed]
- Kastin, A.J.; Akerstrom, V.; Pan, W. Interactions of glucagon-like peptide-1 (GLP-1) with the blood-brain barrier. J. Mol. Neurosci. 2002, 18, 7–14. [Google Scholar] [CrossRef]
- Mietlicki-Baase, E.G.; Ortinski, P.I.; Reiner, D.J.; Sinon, C.G.; McCutcheon, J.E.; Pierce, R.C.; Roitman, M.F.; Hayes, M.R. Glucagon-like peptide-1 receptor activation in the nucleus accumbens core suppresses feeding by increasing glutamatergic ampa/kainate signaling. J. Neurosci. 2014, 34, 6985–6992. [Google Scholar] [CrossRef] [PubMed]
- Orskov, C.; Poulsen, S.S.; Moller, M.; Holst, J.J. Glucagon-like peptide i receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide i. Diabetes 1996, 45, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Punjabi, M.; Arnold, M.; Ruttimann, E.; Graber, M.; Geary, N.; Pacheco-Lopez, G.; Langhans, W. Circulating glucagon-like peptide-1 (GLP-1) inhibits eating in male rats by acting in the hindbrain and without inducing avoidance. Endocrinology 2014, 155, 1690–1699. [Google Scholar] [CrossRef]
- Hayes, M.R.; Kanoski, S.E.; De Jonghe, B.C.; Leichner, T.M.; Alhadeff, A.L.; Fortin, S.M.; Arnold, M.; Langhans, W.; Grill, H.J. The common hepatic branch of the vagus is not required to mediate the glycemic and food intake suppressive effects of glucagon-like-peptide-1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1479–R1485. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ritter, R.C. Circulating GLP-1 and cck-8 reduce food intake by capsaicin-insensitive, nonvagal mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R264–R273. [Google Scholar] [CrossRef]
- Ruttimann, E.B.; Arnold, M.; Hillebrand, J.J.; Geary, N.; Langhans, W. Intrameal hepatic portal and intraperitoneal infusions of glucagon-like peptide-1 reduce spontaneous meal size in the rat via different mechanisms. Endocrinology 2009, 150, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Plamboeck, A.; Veedfald, S.; Deacon, C.F.; Hartmann, B.; Wettergren, A.; Svendsen, L.B.; Meisner, S.; Hovendal, C.; Vilsboll, T.; Knop, F.K.; et al. The effect of exogenous GLP-1 on food intake is lost in male truncally vagotomized subjects with pyloroplasty. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, G1117–G1127. [Google Scholar] [CrossRef] [PubMed]
- Han, V.K.; Hynes, M.A.; Jin, C.; Towle, A.C.; Lauder, J.M.; Lund, P.K. Cellular localization of proglucagon/glucagon-like peptide i messenger RNAs in rat brain. J. Neurosci. Res. 1986, 16, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.J.; Tang-Christensen, M.; Holst, J.J.; Orskov, C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience 1997, 77, 257–270. [Google Scholar] [CrossRef]
- Larsen, P.J.; Tang-Christensen, M.; Jessop, D.S. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. Endocrinology 1997, 138, 4445–4455. [Google Scholar] [CrossRef]
- Zheng, H.; Cai, L.; Rinaman, L. Distribution of glucagon-like peptide 1-immunopositive neurons in human caudal medulla. Brain Struct. Funct. 2015, 220, 1213–1219. [Google Scholar] [CrossRef]
- Lockie, S.H.; Heppner, K.M.; Chaudhary, N.; Chabenne, J.R.; Morgan, D.A.; Veyrat-Durebex, C.; Ananthakrishnan, G.; Rohner-Jeanrenaud, F.; Drucker, D.J.; DiMarchi, R.; et al. Direct control of brown adipose tissue thermogenesis by central nervous system glucagon-like peptide-1 receptor signaling. Diabetes 2012, 61, 2753–2762. [Google Scholar] [CrossRef]
- Kooijman, S.; Wang, Y.; Parlevliet, E.T.; Boon Më, R.; Edelschaap, D.; Snaterse, G.; Pijl, H.; Romijn, J.A.; Rensen, P.C.N. Central GLP-1 receptor signalling accelerates plasma clearance of triacylglycerol and glucose by activating brown adipose tissue in mice. Diabetologia 2015, 58, 2637–2646. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, D.; Gunn, I.; Chen, X.; Bloom, S.; Herbert, J. A role for central glucagon-like peptide-1 in temperature regulation. Neuroreport 1996, 7, 830–832. [Google Scholar] [CrossRef]
- Nogueiras, R.; Diaz-Arteaga, A.; Lockie, S.H.; Velasquez, D.A.; Tschop, J.; Lopez, M.; Cadwell, C.C.; Dieguez, C.; Tschop, M.H. The endocannabinoid system: Role in glucose and energy metabolism. Pharmacol. Res. 2009, 60, 93–98. [Google Scholar] [CrossRef]
- Barrera, J.G.; Jones, K.R.; Herman, J.P.; D’Alessio, D.A.; Woods, S.C.; Seeley, R.J. Hyperphagia and increased fat accumulation in two models of chronic CNS glucagon-like peptide-1 loss of function. J. Neurosci. 2011, 31, 3904–3913. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Hirota, M.; Ohboshi, C.; Shima, K. Identification and localization of glucagon-like peptide-1 and its receptor in rat brain. Endocrinology 1987, 121, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, D.A.; Bagnol, D.; Woods, S.C.; D’Alessio, D.A.; Seeley, R.J. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 2008, 57, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Poleni, P.E.; Akieda-Asai, S.; Koda, S.; Sakurai, M.; Bae, C.R.; Senba, K.; Cha, Y.S.; Furuya, M.; Date, Y. Possible involvement of melanocortin-4-receptor and AMP-activated protein kinase in the interaction of glucagon-like peptide-1 and leptin on feeding in rats. Biochem. Biophys. Res. Commun. 2012, 420, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Merchenthaler, I.; Lane, M.; Shughrue, P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J. Comp. Neurol. 1999, 403, 261–280. [Google Scholar] [CrossRef]
- Vrang, N.; Grove, K. The brainstem preproglucagon system in a non-human primate (macaca mulatta). Brain Res. 2011, 1397, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Beiroa, D.; Imbernon, M.; Gallego, R.; Senra, A.; Herranz, D.; Villaroya, F.; Serrano, M.; Ferno, J.; Salvador, J.; Escalada, J.; et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes 2014, 63, 3346–3358. [Google Scholar] [CrossRef]
- Lopez, M.; Varela, L.; Vazquez, M.J.; Rodriguez-Cuenca, S.; Gonzalez, C.R.; Velagapudi, V.R.; Morgan, D.A.; Schoenmakers, E.; Agassandian, K.; Lage, R.; et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat. Med. 2010, 16, 1001–1008. [Google Scholar] [CrossRef]
- Lopez, M.; Nogueiras, R.; Tena-Sempere, M.; Dieguez, C. Hypothalamic AMPK: A canonical regulator of whole-body energy balance. Nat. Rev. Endocrinol. 2016, 12, 421–432. [Google Scholar] [CrossRef]
- Martinez-Sanchez, N.; Seoane-Collazo, P.; Contreras, C.; Varela, L.; Villarroya, J.; Rial-Pensado, E.; Buque, X.; Aurrekoetxea, I.; Delgado, T.C.; Vazquez-Martinez, R.; et al. Hypothalamic AMPK-er stress-jnk1 axis mediates the central actions of thyroid hormones on energy balance. Cell Metab. 2017, 26, 212–229.e12. [Google Scholar] [CrossRef]
- Lopez, M. AMPK wars: The vmh strikes back, return of the pvh. Trends Endocrinol. Metab. 2018, 29, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Goke, R.; Larsen, P.J.; Mikkelsen, J.D.; Sheikh, S.P. Distribution of GLP-1 binding sites in the rat brain: Evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur. J. Neurosci. 1995, 7, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Reiner, D.J.; Mietlicki-Baase, E.G.; McGrath, L.E.; Zimmer, D.J.; Bence, K.K.; Sousa, G.L.; Konanur, V.R.; Krawczyk, J.; Burk, D.H.; Kanoski, S.E.; et al. Astrocytes regulate GLP-1 receptor-mediated effects on energy balance. J. Neurosci. 2016, 36, 3531–3540. [Google Scholar] [CrossRef] [PubMed]
- Lynch, L.; O’Shea, D.; Winter, D.C.; Geoghegan, J.; Doherty, D.G.; O’Farrelly, C. Invariant NKT cells and cd1d(+) cells amass in human omentum and are depleted in patients with cancer and obesity. Eur. J. Immunol. 2009, 39, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Hogan, A.E.; Tobin, A.M.; Ahern, T.; Corrigan, M.A.; Gaoatswe, G.; Jackson, R.; O’Reilly, V.; Lynch, L.; Doherty, D.G.; Moynagh, P.N.; et al. Glucagon-like peptide-1 (GLP-1) and the regulation of human invariant natural killer t cells: Lessons from obesity, diabetes and psoriasis. Diabetologia 2011, 54, 2745–2754. [Google Scholar] [CrossRef]
- Huh, J.Y.; Kim, J.I.; Park, Y.J.; Hwang, I.J.; Lee, Y.S.; Sohn, J.H.; Lee, S.K.; Alfadda, A.A.; Kim, S.S.; Choi, S.H.; et al. A novel function of adipocytes in lipid antigen presentation to iNKT cells. Mol. Cell Biol. 2013, 33, 328–339. [Google Scholar] [CrossRef]
- Lynch, L.; Nowak, M.; Varghese, B.; Clark, J.; Hogan, A.E.; Toxavidis, V.; Balk, S.P.; O’Shea, D.; O’Farrelly, C.; Exley, M.A. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity 2012, 37, 574–587. [Google Scholar] [CrossRef]
- Lynch, L.; Hogan, A.E.; Duquette, D.; Lester, C.; Banks, A.; LeClair, K.; Cohen, D.E.; Ghosh, A.; Lu, B.; Corrigan, M.; et al. INKT cells induce fgf21 for thermogenesis and are required for maximal weight loss in glp1 therapy. Cell Metab. 2016, 24, 510–519. [Google Scholar] [CrossRef]
- Ji, Y.; Sun, S.; Xia, S.; Yang, L.; Li, X.; Qi, L. Short term high fat diet challenge promotes alternative macrophage polarization in adipose tissue via natural killer t cells and interleukin-4. J. Biol. Chem. 2012, 287, 24378–24386. [Google Scholar] [CrossRef]
- Hui, H.; Farilla, L.; Merkel, P.; Perfetti, R. The short half-life of glucagon-like peptide-1 in plasma does not reflect its long-lasting beneficial effects. Eur. J. Endocrinol. 2002, 146, 863–869. [Google Scholar] [CrossRef]
- Muller, T.D.; Clemmensen, C.; Finan, B.; DiMarchi, R.D.; Tschop, M.H. Anti-obesity therapy: From rainbow pills to polyagonists. Pharmacol. Rev. 2018, 70, 712–746. [Google Scholar] [CrossRef] [PubMed]
- Astrup, A.; Carraro, R.; Finer, N.; Harper, A.; Kunesova, M.; Lean, M.E.; Niskanen, L.; Rasmussen, M.F.; Rissanen, A.; Rossner, S.; et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int. J. Obes. (Lond.) 2012, 36, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Wadden, T.A.; Hollander, P.; Klein, S.; Niswender, K.; Woo, V.; Hale, P.M.; Aronne, L. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: The scale maintenance randomized study. Int. J. Obes. (Lond.) 2015, 39, 187. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.; Acosta, A. GLP-1 receptor agonists: Nonglycemic clinical effects in weight loss and beyond. Obesity (Silver Spring) 2015, 23, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.J.; Gotz, A.; Tschop, M.H.; Muller, T.D. Gut hormone polyagonists for the treatment of type 2 diabetes. Peptides 2018, 100, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Finan, B.; Clemmensen, C.; DiMarchi, R.D.; Tschop, M.H. The new biology and pharmacology of glucagon. Physiol. Rev. 2017, 97, 721–766. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.J.; Konkar, A.; Hornigold, D.C.; Trevaskis, J.L.; Jackson, R.; Fritsch Fredin, M.; Jansson-Lofmark, R.; Naylor, J.; Rossi, A.; Bednarek, M.A.; et al. Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes. Metab. 2016, 18, 1176–1190. [Google Scholar] [CrossRef]
- Finan, B.; Clemmensen, C.; Muller, T.D. Emerging opportunities for the treatment of metabolic diseases: Glucagon-like peptide-1 based multi-agonists. Mol. Cell Endocrinol. 2015, 418 Pt 1, 42–54. [Google Scholar] [CrossRef]
- Clark, A.; Saad, M.F.; Nezzer, T.; Uren, C.; Knowler, W.C.; Bennett, P.H.; Turner, R.C. Islet amyloid polypeptide in diabetic and non-diabetic pima indians. Diabetologia 1990, 33, 285–289. [Google Scholar] [CrossRef]
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1219–1235. [Google Scholar] [CrossRef] [Green Version]
- Lutz, T.A. Roles of amylin in satiation, adiposity and brain development. Forum. Nutr. 2010, 63, 64–74. [Google Scholar] [PubMed]
- Lutz, T.A. The role of amylin in the control of energy homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1475–R1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, W.T.; Balasubramaniam, A.; Zhang, F.S.; Wimalawansa, S.J.; Fischer, J.E. Anorexia following the intrahypothalamic administration of amylin. Brain Res. 1991, 539, 352–354. [Google Scholar] [CrossRef]
- Trevaskis, J.L.; Mack, C.M.; Sun, C.; Soares, C.J.; D’Souza, L.J.; Levy, O.E.; Lewis, D.Y.; Jodka, C.M.; Tatarkiewicz, K.; Gedulin, B.; et al. Improved glucose control and reduced body weight in rodents with dual mechanism of action peptide hybrids. PLoS ONE 2013, 8, e78154. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Trevaskis, J.L.; Jodka, C.M.; Neravetla, S.; Griffin, P.; Xu, K.; Wang, Y.; Parkes, D.G.; Forood, B.; Ghosh, S.S. Bifunctional pegylated exenatide-amylinomimetic hybrids to treat metabolic disorders: An example of long-acting dual hormonal therapeutics. J. Med. Chem. 2013, 56, 9328–9341. [Google Scholar] [CrossRef] [PubMed]
- Finan, B.; Ma, T.; Ottaway, N.; Muller, T.D.; Habegger, K.M.; Heppner, K.M.; Kirchner, H.; Holland, J.; Hembree, J.; Raver, C.; et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci. Transl. Med. 2013, 5, 209ra151. [Google Scholar] [CrossRef]
- Dupre, J.; Ross, S.A.; Watson, D.; Brown, J.C. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J. Clin. Endocrinol. Metab. 1973, 37, 826–828. [Google Scholar] [CrossRef]
- Christensen, M.; Vedtofte, L.; Holst, J.J.; Vilsboll, T.; Knop, F.K. Glucose-dependent insulinotropic polypeptide: A bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes 2011, 60, 3103–3109. [Google Scholar] [CrossRef]
- Christensen, M.B.; Calanna, S.; Holst, J.J.; Vilsboll, T.; Knop, F.K. Glucose-dependent insulinotropic polypeptide: Blood glucose stabilizing effects in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E418–E426. [Google Scholar] [CrossRef]
- Martin, C.M.; Irwin, N.; Flatt, P.R.; Gault, V.A. A novel acylated form of (d-ala(2))gip with improved antidiabetic potential, lacking effect on body fat stores. Biochim. Biophys. Acta 2013, 1830, 3407–3413. [Google Scholar] [CrossRef]
- Finan, B.; Yang, B.; Ottaway, N.; Smiley, D.L.; Ma, T.; Clemmensen, C.; Chabenne, J.; Zhang, L.; Habegger, K.M.; Fischer, K.; et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015, 21, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Jall, S.; Sachs, S.; Clemmensen, C.; Finan, B.; Neff, F.; DiMarchi, R.D.; Tschop, M.H.; Muller, T.D.; Hofmann, S.M. Monomeric GLP-1/gip/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol. Metab. 2017, 6, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Finan, B.; Yang, B.; Ottaway, N.; Stemmer, K.; Muller, T.D.; Yi, C.X.; Habegger, K.; Schriever, S.C.; Garcia-Caceres, C.; Kabra, D.G.; et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med. 2012, 18, 1847–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finan, B.; Clemmensen, C.; Zhu, Z.; Stemmer, K.; Gauthier, K.; Muller, L.; De Angelis, M.; Moreth, K.; Neff, F.; Perez-Tilve, D.; et al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell 2016, 167, 843–857.e14. [Google Scholar] [CrossRef] [PubMed]
- Quarta, C.; Clemmensen, C.; Zhu, Z.; Yang, B.; Joseph, S.S.; Lutter, D.; Yi, C.X.; Graf, E.; Garcia-Caceres, C.; Legutko, B.; et al. Molecular integration of incretin and glucocorticoid action reverses immunometabolic dysfunction and obesity. Cell Metab. 2017, 26, 620–632.e6. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Crespo, M.; Csikasz, R.I.; Martinez-Sanchez, N.; Dieguez, C.; Cannon, B.; Nedergaard, J.; Lopez, M. Essential role of ucp1 modulating the central effects of thyroid hormones on energy balance. Mol. Metab. 2016, 5, 271–282. [Google Scholar] [CrossRef]
- Bianco, A.C.; Sheng, X.Y.; Silva, J.E. Triiodothyronine amplifies norepinephrine stimulation of uncoupling protein gene transcription by a mechanism not requiring protein synthesis. J. Biol. Chem. 1988, 263, 18168–18175. [Google Scholar]
- Martinez-Sanchez, N.; Moreno-Navarrete, J.M.; Contreras, C.; Rial-Pensado, E.; Ferno, J.; Nogueiras, R.; Dieguez, C.; Fernandez-Real, J.M.; Lopez, M. Thyroid hormones induce browning of white fat. J. Endocrinol. 2017, 232, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Colman, E. Anorectics on trial: A half century of federal regulation of prescription appetite suppressants. Ann. Intern. Med. 2005, 143, 380–385. [Google Scholar] [CrossRef]
- Kuo, D.Y.; Chen, P.N.; Yu, C.H.; Kuo, M.H.; Hsieh, Y.S.; Chu, S.C. Involvement of neuropeptide y y1 receptor in the regulation of amphetamine-mediated appetite suppression. Neuropharmacology 2012, 63, 842–850. [Google Scholar] [CrossRef]
- Kuo, D.Y.; Yang, S.F.; Chu, S.C.; Chu, S.C.; Chen, C.H.; Hsieh, Y.S. Amphetamine-evoked changes of oxidative stress and neuropeptide y gene expression in hypothalamus: Regulation by the protein kinase c-delta signaling. Chem. Biol. Interact. 2009, 180, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Heisler, L.K.; Cowley, M.A.; Tecott, L.H.; Fan, W.; Low, M.J.; Smart, J.L.; Rubinstein, M.; Tatro, J.B.; Marcus, J.N.; Holstege, H.; et al. Activation of central melanocortin pathways by fenfluramine. Science 2002, 297, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Heisler, L.K.; Jobst, E.E.; Sutton, G.M.; Zhou, L.; Borok, E.; Thornton-Jones, Z.; Liu, H.Y.; Zigman, J.M.; Balthasar, N.; Kishi, T.; et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron 2006, 51, 239–249. [Google Scholar] [CrossRef] [PubMed]
- McDuffie, J.R.; Calis, K.A.; Booth, S.L.; Uwaifo, G.I.; Yanovski, J.A. Effects of orlistat on fat-soluble vitamins in obese adolescents. Pharmacotherapy 2002, 22, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Melia, A.T.; Koss-Twardy, S.G.; Zhi, J. The effect of orlistat, an inhibitor of dietary fat absorption, on the absorption of vitamins a and e in healthy volunteers. J. Clin. Pharmacol. 1996, 36, 647–653. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-García, I.; Milbank, E.; Diéguez, C.; López, M.; Contreras, C. Glucagon, GLP-1 and Thermogenesis. Int. J. Mol. Sci. 2019, 20, 3445. https://doi.org/10.3390/ijms20143445
González-García I, Milbank E, Diéguez C, López M, Contreras C. Glucagon, GLP-1 and Thermogenesis. International Journal of Molecular Sciences. 2019; 20(14):3445. https://doi.org/10.3390/ijms20143445
Chicago/Turabian StyleGonzález-García, Ismael, Edward Milbank, Carlos Diéguez, Miguel López, and Cristina Contreras. 2019. "Glucagon, GLP-1 and Thermogenesis" International Journal of Molecular Sciences 20, no. 14: 3445. https://doi.org/10.3390/ijms20143445
APA StyleGonzález-García, I., Milbank, E., Diéguez, C., López, M., & Contreras, C. (2019). Glucagon, GLP-1 and Thermogenesis. International Journal of Molecular Sciences, 20(14), 3445. https://doi.org/10.3390/ijms20143445