Development and Applications of Superfolder and Split Fluorescent Protein Detection Systems in Biology


Abstract
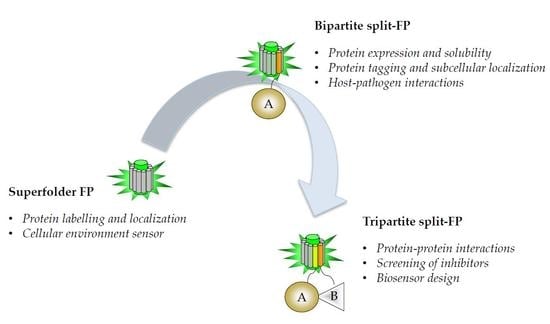
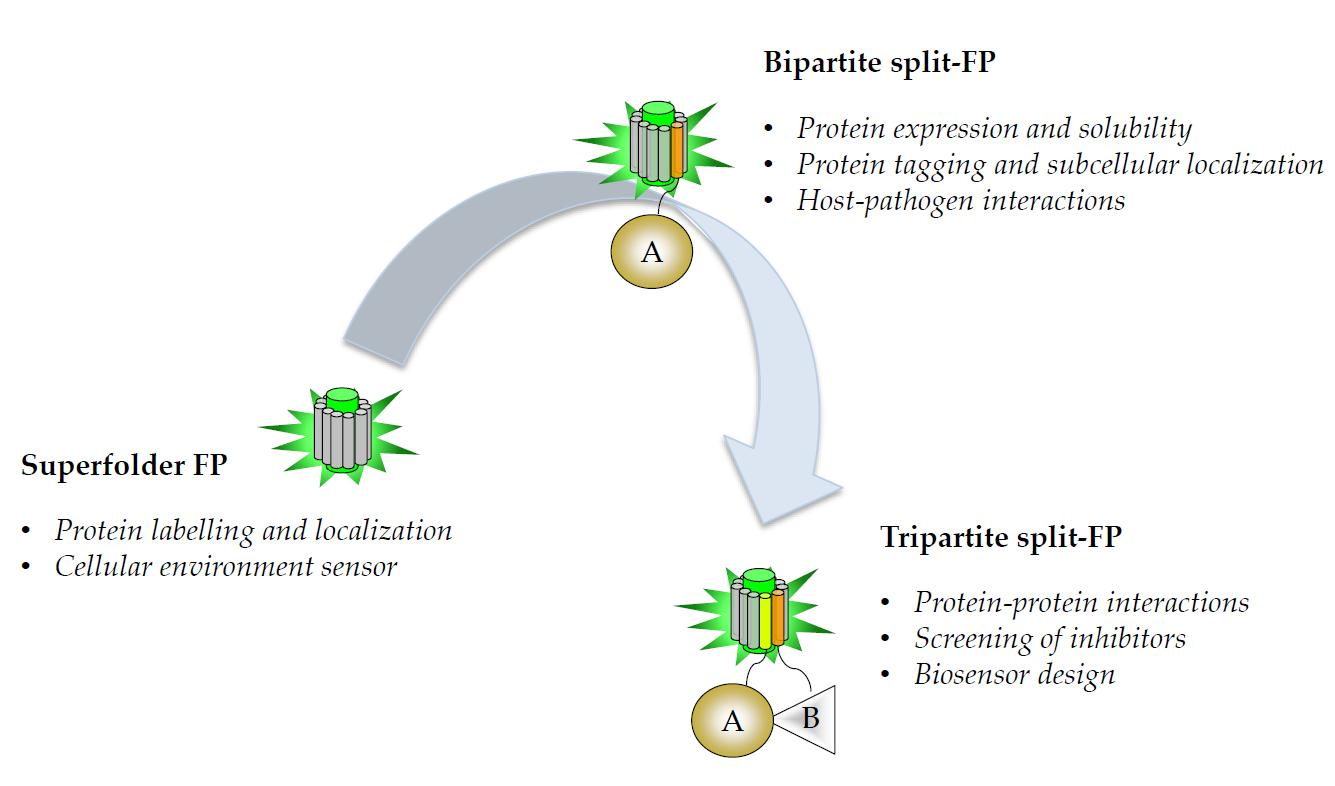{kind=link}
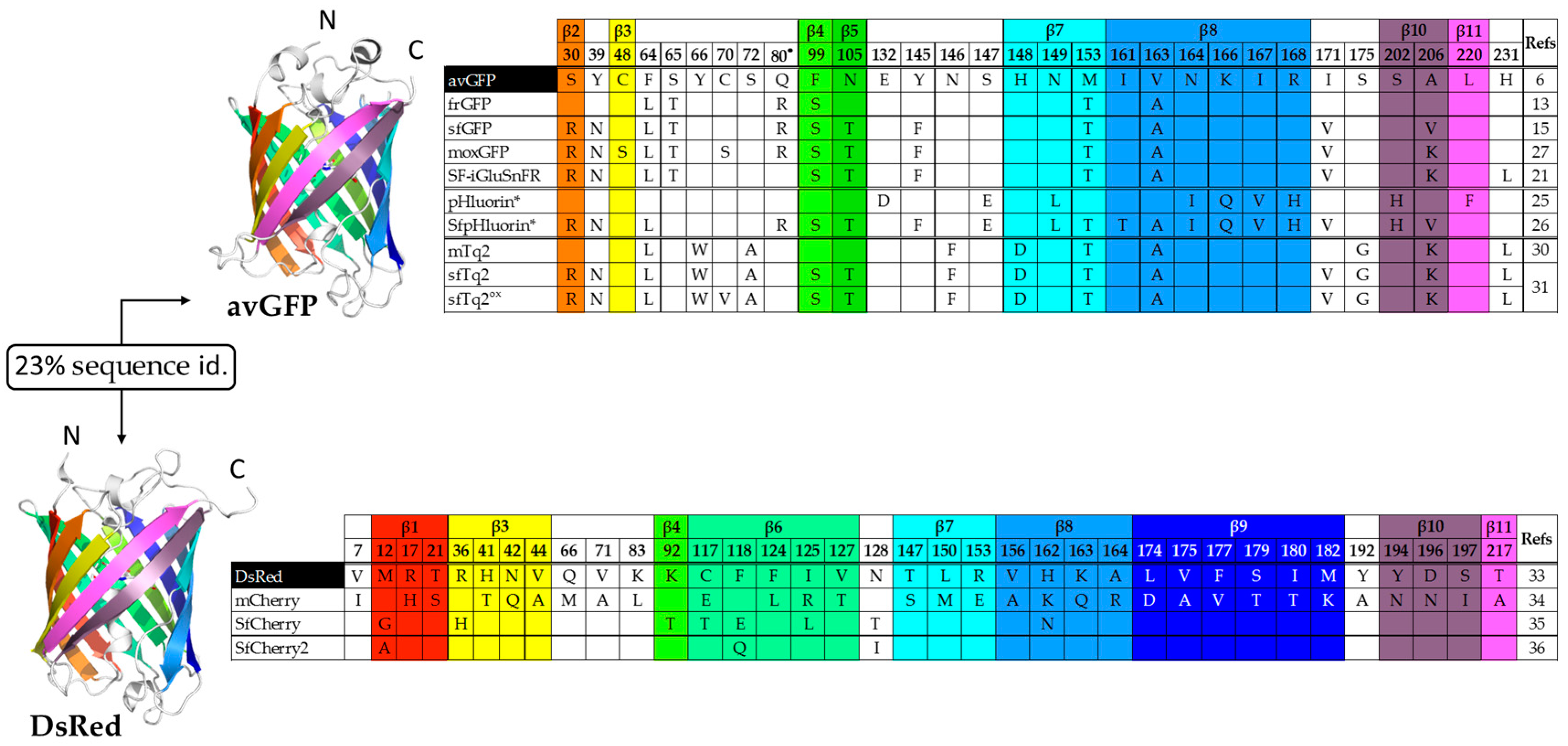{kind=link}
{kind=link}
{kind=link}
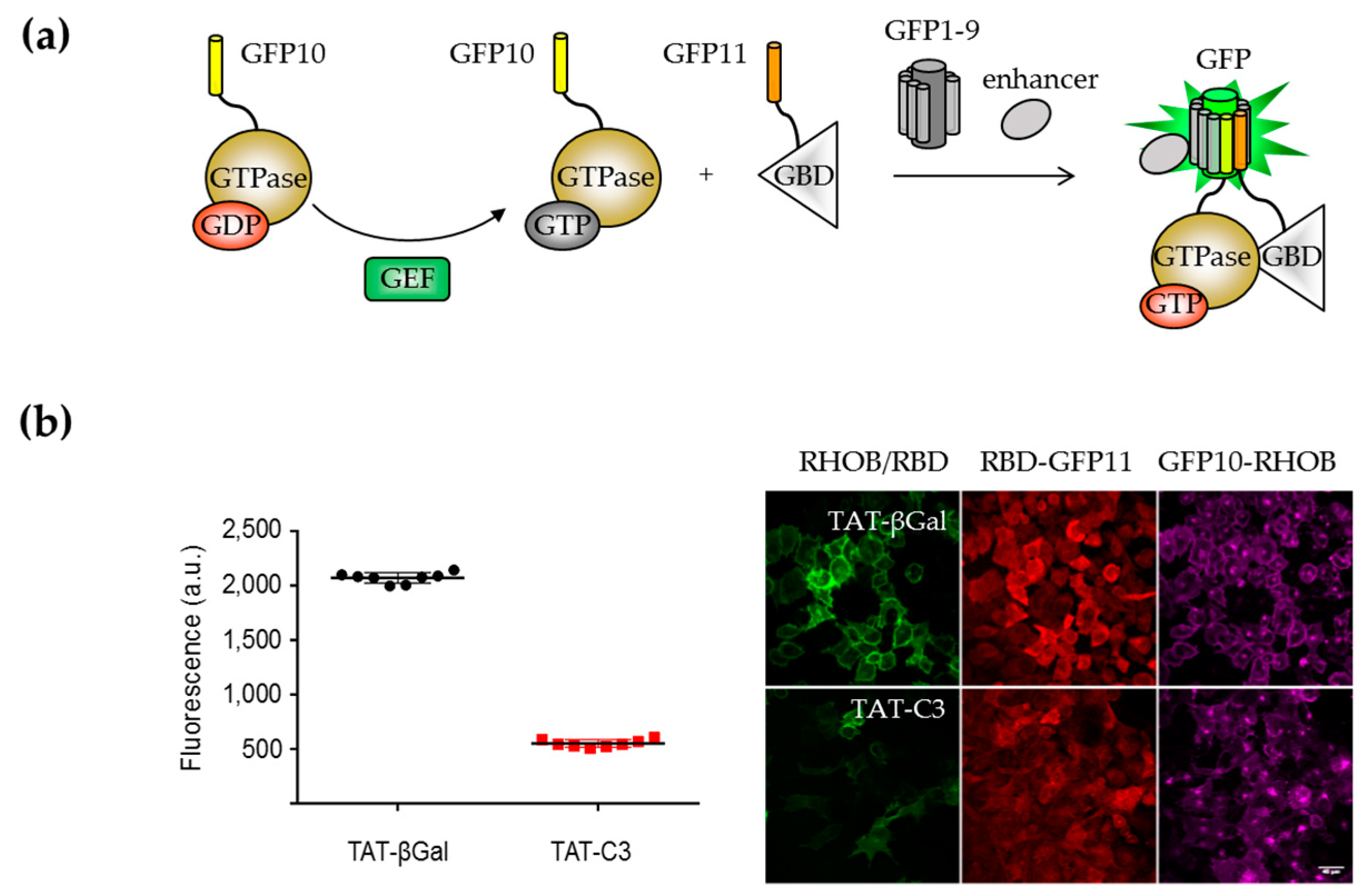{kind=link}
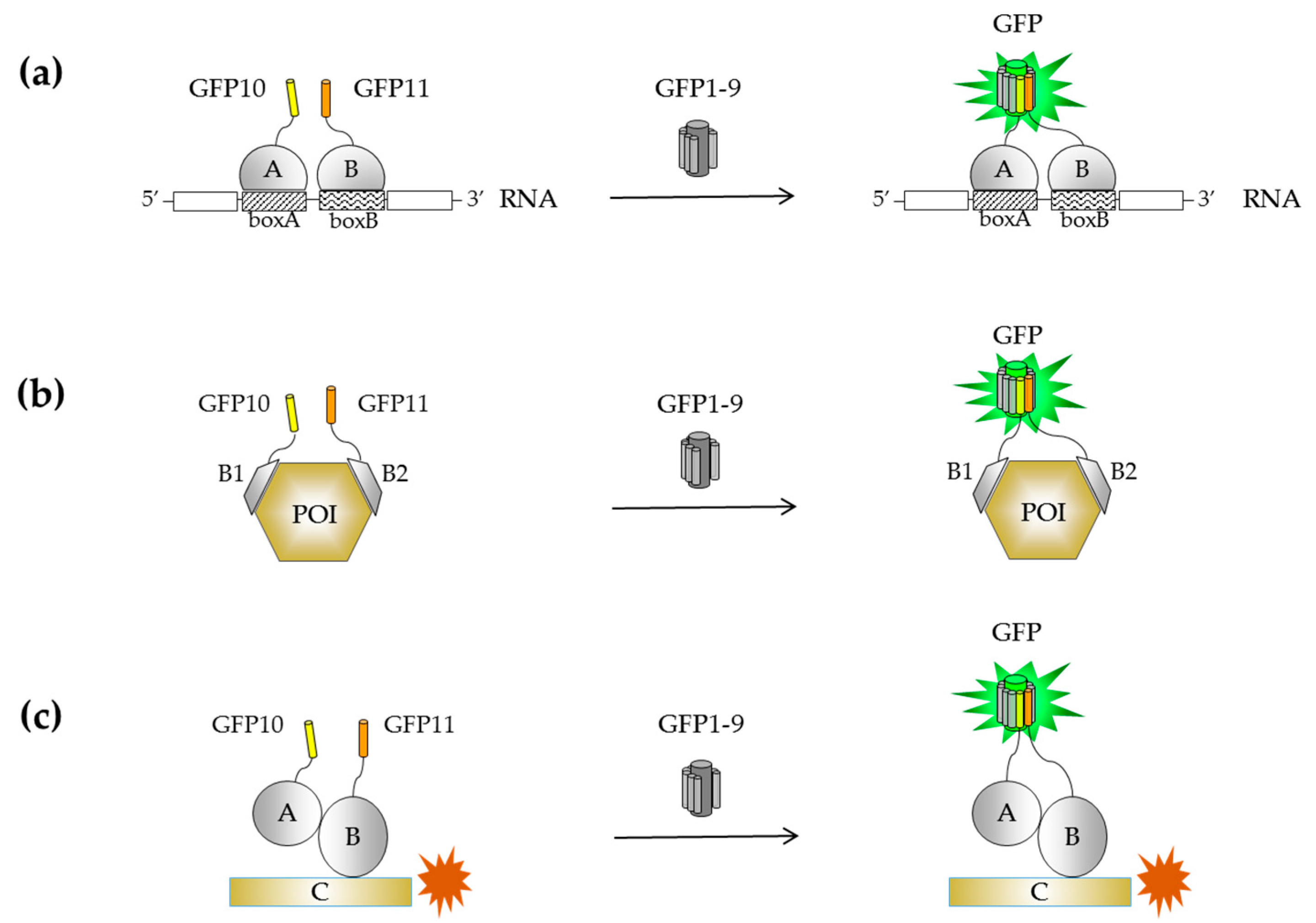{kind=link}
1. Superfolder Fluorescent Proteins: Progenitor of Split Fluorescent Protein (FP) Systems
1.1. Improving the Folding of avGFP
1.2. Superfolder Color Variants and Applications
2. Split-Fluorescent Proteins
2.1. Self-Associating Split-FP Systems
2.1.1. Engineering of the Bipartite GFP1–10/GFP11 System
2.1.2. Applications of the Bipartite Split-GFP System to Monitor Protein Solubility In Vivo and In Vitro
2.1.3. Adapting the GFP1–10/GFP11 System for Protein Labelling in Mammalian Cells
2.1.4. Applications in Cell Biology
Neuronal Cell Communication
Protein Topology and Subcellular Localization
Host–Pathogen Interactions
2.2. Protein–Protein Interactions
2.2.1. Engineering of a Tripartite Split-GFP System
2.2.2. Known Examples of Tripartite Split-GFP Applications in Cell Biology
Screening of Novel Inhibitors
Monitoring Direct Associations
Monitoring Indirect Associations
Biosensor Design
3. What’s Next in the Split-FP Development?
3.1. Extending the Spectra of Split Fluorescent Protein Detection Systems
3.2. Improving the Maturation Rate of the Chromophore After Complementation
3.2.1. Engineering of Detection Fragments for Improved Conformational Homogeneity
3.2.2. Prematuration of the Chromophore for In Vitro Assays
3.2.3. Use of Scaffolds Binders to Increase the Stability of the Chromophore
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASAP1 | Accelerated Sensor of Action Potentials |
| AvGFP | Aequorea victoria Green Fluorescent Protein |
| BiFC | Bimolecular Fluorescence Complementation |
| CALM | Complementation-Activated Light Microscopy |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
| ER | Endoplasmic Reticulum |
| FKBP | FK506 Binding Protein |
| FP | Fluorescent Protein |
| FRAP | Fluorescent Recovery After Photobleaching |
| FRB | FKBP-Rapamycin Binding domain |
| FRET | Förster Resonance Energy Transfer |
| GBD | GTPase Binding Domain |
| GEF | Guanine nucleotide Exchange Factor |
| ORF | Open Reading Frame |
| PCA | Protein Complementation Assay |
| POI | Protein Of Interest |
| PPI | Protein-Protein Interaction |
| RAGE | Receptor for Advanced Glycation End product |
| RAP | Rapamycin |
| RBD | RHO Binding Domain |
| SPR | Surface Plasmon Resonance |
| SRTA | Sortase A |
| TetFC | Tetramolecular Fluorescence Complementation |
| TFIIH | Transcription Factor II H |
| TTDA | TrichoThioDystrophy-A |
| Tth | Thermus thermophiles |
| TriFC | Trimolecular Fluorescence Complementation |
| Y2H | Yeast-two-hybrid |
References
- Rodriguez, E.A.; Campbell, R.E.; Lin, J.Y.; Lin, M.Z.; Miyawaki, A.; Palmer, A.E.; Shu, X.; Zhang, J.; Tsien, R.Y. The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci. 2017, 42, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Chudakov, D.; Matz, M.; Lukyanov, S.; Lukyanov, K. Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues. Physiol. Rev. 2010, 90, 1103–1163. [Google Scholar] [CrossRef] [PubMed]
- Kremers, G.-J.; Gilbert, S.G.; Cranfill, P.J.; Davidson, M.W.; Piston, D.W. Fluorescent proteins at a glance. J. Cell Sci. 2011, 124, 2676. [Google Scholar] [CrossRef][Green Version]
- Romei, M.G.; Boxer, S.G. Split Green Fluorescent Proteins: Scope, Limitations, and Outlook. Annu. Rev. Biophys. 2019, 48, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Lambert, T.J. FPbase: A community-editable fluorescent protein database. Nat. Methods 2019, 16, 277–278. [Google Scholar] [CrossRef]
- Prasher, D.C.; Eckenrode, V.K.; Ward, W.W.; Prendergast, F.G.; Cormier, M.J. Primary structure of the Aequorea victoria green-fluorescent protein. Gene 1992, 111, 229–233. [Google Scholar] [CrossRef]
- Tsien, R.Y. The Green Fluorescent Protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Cubitt, A.B.; Heim, R.; Adams, S.R.; Boyd, A.E.; Gross, L.A.; Tsien, R.Y. Understanding, improving and using green fluorescent proteins. Trends Biochem. Sci. 1995, 20, 448–455. [Google Scholar] [CrossRef]
- Heim, R.; Prashert, D.C.; Tsien, R.Y. Wavelength mutations and posttranslational autoxidation of green fluorescent protein. Proc. Natl. Acad. Sci. USA 1994, 91, 12501–12504. [Google Scholar] [CrossRef]
- Chattoraj, M.; King, B.A.; Bublitz, G.U.; Boxert, S.G. Ultra-fast excited state dynamics in green fluorescent protein: Multiple states and proton transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 8362–8367. [Google Scholar] [CrossRef]
- Heim, R.; Cubitt, A.B.; Tsien, R.Y. Improved green fluorescence. Nature 1995, 373, 663–664. [Google Scholar] [CrossRef] [PubMed]
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38. [Google Scholar] [CrossRef]
- Waldo, G.S.; Standish, B.M.; Berendzen, J.; Terwilliger, T.C. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 1999, 17, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Crameri, A.; Whitehorn, E.A.; Tate, E.; Stemmer, W.P.C. Improved Green Fluorescent Protein by Molecular Evolution Using DNA Shuffling. Nat. Biotechnol. 1996, 14, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Pedelacq, J.-D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Nagai, T.; Ibata, K.; Park, E.S.; Kubota, M.; Mikoshiba, K. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat. Biotechnol. 2001, 20, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C. Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science 2002, 296, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Patterson, G.H.; Davidson, M.W. Advances in fluorescent protein technology. J. Cell Sci. 2007, 4247–4260. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green Fluorescent Protein as a Marker for Gene Expression. Science 1994, 263, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Cava, F.; de Pedro, M.A.; Blas-galindo, E.; Waldo, G.S.; Westblade, L.F.; Berenguer, J. Expression and use of superfolder green fluorescent protein at high temperatures in vivo: A tool to study extreme thermophile biology. Environ. Microbiol. 2008, 10, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Marvin, J.S.; Scholl, B.; Wilson, D.E.; Podgorski, K.; Kazemipour, A.; Müller, J.A.; Schoch, S.; José, F.; Quiroz, U.; Rebola, N.; et al. Stability, affinity, and chromatic variants of the glutamate sensor iGluSnFR. Nat. Methods 2018, 15, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Lobas, M.A.; Tao, R.; Nagai, J.; Kronschläger, M.T.; Marvin, J.S.; Looger, L.L.; Khakh, B.S.; Borden, P.M. A genetically encoded single-wavelength sensor for imaging cytosolic and cell surface ATP. Nat. Commun. 2019. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pierre, F.; Marshall, J.D.; Yang, Y.; Gong, Y.; Schnitzer, M.J.; Lin, M.Z. High-fidelity optical reporting of neuronal electrical activity with an ultrafast fluorescent voltage sensor. Nat. Neurosci. 2014, 17, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Morciano, G.; Sarti, A.C.; Marchi, S.; Missiroli, S.; Falzoni, S.; Raffaghello, L.; Pistoia, V.; Giorgi, C.; di Virgilio, F.; Pinton, P. Use of luciferase probes to measure ATP in living cells and animals. Nat. Protoc. 2017, 12, 1542–1562. [Google Scholar] [CrossRef] [PubMed]
- Miesenbock, G.; de Angelis, D.A.; Rothman, J.E. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 1998, 394, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Reifenrath, M.; Boles, E. A superfolder variant of pH- sensitive pHluorin for in vivo pH measurements in the endoplasmic reticulum. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Costantini, L.M.; Baloban, M.; Markwardt, M.L.; Rizzo, M.; Guo, F.; Verkhusha, V.V.; Snapp, E.L. A palette of fluorescent proteins optimized for diverse cellular environments. Nat. Commun. 2015, 6, 7670. [Google Scholar] [CrossRef]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.G.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef]
- Aronson, D.E.; Costantini, L.M.; Snapp, E.L. Superfolder GFP Is Fluorescent in Oxidizing Environments When Targeted via the Sec Translocon. Traffic 2011, 12, 543–548. [Google Scholar] [CrossRef]
- Goedhart, J.; von Stetten, D.; Noirclerc-savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W.J., Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef]
- Meiresonne, N.Y.; Consoli, E.; Mertens, L.M.Y.; Chertkova, A.O.; Goedhart, J.; Den Blaauwen, T. Superfolder mTurquoise2 ox optimized for the bacterial periplasm allows high efficiency in vivo FRET of cell division antibiotic targets. Mol. Microbiol. 2019, 111, 1025–1038. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Shaner, N.C.; Yarbrough, C.A.; Tsien, R.Y.; Remington, S.J. Novel Chromophores and Buried Charges Control Color in mFruits. Biochemistry 2006, 45. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.B.; Hung, L.W.; Yeates, T.O.; Terwilliger, T.C.; Waldo, G.S. Split green fluorescent protein as a modular binding partner for protein crystallization. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2513–2523. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Sekine, S.; Pessino, V.; Li, H.; Leonetti, M.D.; Huang, B. Improved split fluorescent proteins for endogenous protein labeling. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Dennig, A.; Shivange, A.V.; Marienhagen, J.; Schwaneberg, U. OmniChange: The Sequence Independent Method for Simultaneous Site-Saturation of Five Codons. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Bindels, D.S.; Haarbosch, L.; van Weeren, L.; Postma, M.; Wiese, K.E.; Mastop, M.; Aumonier, S.; Gotthard, G.; Royant, A.; Hink, M.A.; et al. mScarlet: A bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 2016, 14, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef]
- Cabantous, S.; Waldo, G.S. In vivo and in vitro protein solubility assays using split GFP. Nat. Methods 2006, 3, 845–854. [Google Scholar] [CrossRef]
- Listwan, P.; Pédelacq, J.D.; Lockard, M.; Bell, C.; Terwilliger, T.C.; Waldo, G.S. The optimization of in vitro high-throughput chemical lysis of Escherichia coli. Application to ACP domain of the polyketide synthase ppsC from Mycobacterium tuberculosis. J. Struct. Funct. Genom. 2010, 11, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Pédelacq, J.D.; Mark, B.L.; Naranjo, C.; Terwilliger, T.C.; Waldo, G.S. Recent advances in GFP folding reporter and split-GFP solubility reporter technologies. Application to improving the folding and solubility of recalcitrant proteins from Mycobacterium tuberculosis. J. Struct. Funct. Genom. 2005, 6, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Rottier, K.; Faille, A.; Prudhomme, T.; Leblanc, C.; Chalut, C.; Cabantous, S.; Guilhot, C.; Mourey, L.; Pedelacq, J.D. Detection of soluble co-factor dependent protein expression in vivo: Application to the 4′-phosphopantetheinyl transferase PptT from Mycobacterium tuberculosis. J. Struct. Biol. 2013, 183, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Santos-Aberturas, J.; Dörr, M.; Bornscheuer, U.T. Normalized screening of protein engineering libraries by split-GFP crude cell extract quantification. Methods Mol. Biol. 2018, 1685, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Pedelacq, J.-D.; Nguyen, H.B.; Cabantous, S.; Mark, B.L.; Listwan, P.; Bell, C.; Friedland, N.; Lockard, M.; Faille, A.; Mourey, L.; et al. Experimental mapping of soluble protein domains using a hierarchical approach. Nucleic Acids Res. 2011, 39, e125. [Google Scholar] [CrossRef] [PubMed]
- Massemin, A.; Cabantous, S.; Waldo, G.S.; Pedelacq, J.-D. High-Throughput Isolation of Soluble Protein Domains Using a Bipartite Split-GFP Complementation System. Methods Mol. Biol. 2019, 2025, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Chun, W.; Waldo, G.S.; Johnson, G.V.W. Split GFP complementation assay: A novel approach to quantitatively measure aggregation of tau in situ: Effects of GSK3 b activation and caspase 3 cleavage. J. Neurochem. 2007, 103, 2529–2539. [Google Scholar] [CrossRef]
- Kothawala, A.; Kilpatrick, K.; Novoa, J.A.; Segatori, L. Quantitative Analysis of a-Synuclein Solubility in Living Cells Using Split GFP Complementation. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Kaddoum, L.; Magdeleine, E.; Waldo, G.S.; Joly, E.; Cabantous, S. One-step split GFP staining for sensitive protein detection and localization in mammalian cells. BioTechniques 2010, 49. [Google Scholar] [CrossRef]
- Pinaud, F.; Dahan, M. Targeting and imaging single biomolecules in living cells by complementation-activated light microscopy with split- fl uorescent proteins. Proc. Natl. Acad. Sci. USA 2011, 108, 201–210. [Google Scholar] [CrossRef]
- Kamiyama, D.; Sekine, S.; Barsi-rhyne, B.; Hu, J.; Chen, B.; Gilbert, L.A.; Ishikawa, H.; Leonetti, M.D.; Marshall, W.F.; Weissman, J.S.; et al. Versatile protein tagging in cells with split fluorescent protein. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, M.D.; Sekine, S.; Kamiyama, D.; Weissman, J.S.; Huang, B. A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proc. Natl. Acad. Sci. USA 2016, 3501–3508. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, E.H.; Vanhoven, M.K.; Bendesky, A.; Wang, G.; Fetter, R.D.; Shen, K.; Bargmann, C.I. Neurotechnique GFP Reconstitution Across Synaptic Partners ( GRASP ) Defines Cell Contacts and Synapses in Living Nervous Systems. Neuron 2008, 57, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Manoli, D.S.; Ahmed, O.M.; Chen, Y.; Agarwal, N.; Kwong, S.; Cai, A.G.; Neitz, J.; Renslo, A.; Baker, B.S.; et al. Genetic and Neural Mechanisms that Inhibit Drosophila from Mating with Other Species. Cell 2013, 154, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Gordon, M.D.; Scott, K. Article Motor Control in a Drosophila Taste Circuit. Neuron 2009, 61, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, M.; Sanes, J.R. Transgenic strategy for identifying synaptic connections in mice by fluorescence complementation (GRASP). Front. Mol. Neurosci. 2012, 5. [Google Scholar] [CrossRef]
- Macpherson, L.J.; Zaharieva, E.E.; Kearney, P.J.; Alpert, M.H.; Lin, T.; Turan, Z.; Lee, C.; Gallio, M. Dynamic labelling of neural connections in multiple colours by trans-synaptic fluorescence complementation. Nat. Commu. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hyun, S.; Maruri-avidal, L.; Moss, B. Topology of Endoplasmic Reticulum-Associated Cellular and Viral Proteins Determined with Split-GFP. Traffic 2015, 787–795. [Google Scholar] [CrossRef]
- Zhong, Y.; Fang, S. Live Cell Imaging of Protein Dislocation from the Endoplasmic Reticulum. J. Biol. Chem. 2012, 287, 28057–28066. [Google Scholar] [CrossRef]
- Zhang, B.; Rapolu, M.; Liang, Z.; Han, Z.; Williams, P.G.; Su, W.W. A Dual-Intein Autoprocessing A Dual-Intein Autoprocessing Domain that Directs Synchronized Protein Co-Expression in Both Prokaryotes and Eukaryotes. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Cali, T.; Ottolini, D.; Soriano, M.E.; Brini, M. A new split-GFP-based probe reveals DJ-1 translocation into the mitochondrial matrix to sustain ATP synthesis upon nutrient deprivation. Hum. Mol. Genet. 2015, 24, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Cieri, D.; Vicario, M.; Giacomello, M.; Vallese, F.; Filadi, R.; Wagner, T.; Pozzan, T.; Pizzo, P.; Scorrano, L.; Brini, M.; et al. SPLICS: A split green fl uorescent protein-based contact site sensor for narrow and wide heterotypic organelle juxtaposition. Cell Death Differ. 2018, 2, 1131–1145. [Google Scholar] [CrossRef] [PubMed]
- Kakimoto, Y.; Tashiro, S.; Kojima, R.; Morozumi, Y.; Endo, T. Visualizing multiple inter-organelle contact sites using the organelle- targeted split-GFP system. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, X.; Xu, J.; Shang, W.; Tong, C. A novel fluorescent reporter detects plastic remodeling of mitochondria–ER contact sites. J. Cell Sci. 2018, 131, jcs208686. [Google Scholar] [CrossRef] [PubMed]
- Smoyer, C.J.; Katta, S.S.; Gardner, J.M.; Stoltz, L.; Mccroskey, S.; Bradford, W.D.; Mcclain, M.; Smith, S.E.; Slaughter, B.D.; Unruh, J.R.; et al. Analysis of membrane proteins localizing to the inner nuclear envelope in living cells. J. Cell Biol. 2016, 215, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Nielsen, M.E.; Pedersen, C.; Thordal-christensen, H. A Split-GFP Gateway Cloning System for Topology Analyses of Membrane Proteins in Plants. PLoS ONE 2017. [Google Scholar] [CrossRef]
- Avilov, S.V.; Moisy, D.; Munier, S.; Schraidt, O.; Naffakh, N.; Cusack, S. Replication-Competent Influenza A Virus That Encodes a Split-Green Live-Cell Imaging of the Virus Life Cycle. J. Virol. 2012, 86, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Van Engelenburg, S.B.; Palmer, A.E. Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat. Methods 2010, 7, 325–333. [Google Scholar] [CrossRef]
- Külzer, S.; Petersen, W.; Baser, A.; Mandel, K.; Przyborski, J.M. Molecular & Biochemical Parasitology Use of self-assembling GFP to determine protein topology and compartmentalisation in the Plasmodium falciparum -infected erythrocyte. Mol. Biochem. Parasitol. 2013, 187, 87–90. [Google Scholar] [CrossRef]
- Avilov, S.V.; Aleksandrova, N. Fluorescence protein complementation in microscopy: Applications beyond detecting bi-molecular interactions. Methods Appl. Fluoresc. 2018, 7, 012001. [Google Scholar] [CrossRef]
- Young, A.M.; Minson, M.; Mcquate, S.E.; Palmer, A.E. Optimized Fluorescence Complementation Platform for Visualizing Salmonella Effector Proteins Reveals Distinctly Different Intracellular Niches in Different Cell Types. ACS Infect. Dis. 2017, 3, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Batan, D.; Braselmann, E.; Minson, M.; My, D.; Nguyen, T.; Cossart, P.; Palmer, A.E. A Multicolor Split-Fluorescent Protein Approach to Visualize Listeria Protein Secretion in Infection. Biophys. J. 2018, 114, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, Q.; Tu, H.; Lim, Z.; Pan, S.Q. Direct visualization of Agrobacterium-delivered VirE2 in recipient cells. Plant J. 2014, 77, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, X.; Tu, H.; Pan, S.Q. Agrobacterium-delivered virulence protein VirE2 is trafficked inside host cells via a myosin XI-K–powered ER/actin network. Proc. Natl. Acad. Sci. USA 2017, 114, 2982–2987. [Google Scholar] [CrossRef] [PubMed]
- Lievens, S.; Lemmens, I.; Tavernier, J. Mammalian two-hybrids come of age. Trends Biochem. Sci. 2009, 34, 579–588. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brückner, A.; Polge, C.; Lentze, N.; Auerbach, D.; Schlattner, U. Yeast Two-Hybrid, a Powerful Tool for Systems Biology. Int. J. Mol. Sci. 2009, 10, 2763–2788. [Google Scholar] [CrossRef]
- Wilson, C.G.M.; Magliery, T.J.; Regan, L. Detecting protein-protein interactions with GFP-fragment reassembly. Nat. Methods 2004, 1, 255–262. [Google Scholar] [CrossRef]
- Ghosh, I.; Hamilton, A.D.; Regan, L. Antiparallel Leucine Zipper-Directed Protein Reassembly: Application to the Green Fluorescent Protein. J. Am. Chem. Soc. 2000, 122, 5658–5659. [Google Scholar] [CrossRef]
- Hu, C.; Kerppola, T.K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 2003, 21, 539–545. [Google Scholar] [CrossRef]
- Bischof, J.; Duffraisse, M.; Furger, E.; Ajuria, L.; Giraud, G.; Vanderperre, S.; Paul, R.; Bjo, M.; Ahr, D.; Ahmed, A.W.; et al. Generation of a versatile BiFC ORFeome library for analyzing protein–protein interactions in live Drosophila. Elife 2018, 7, e38853. [Google Scholar] [CrossRef]
- Wiens, M.D.; Campbell, R.E. Surveying the landscape of optogenetic methods for detection of protein–protein interactions. Syst. Biol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Morell, M.; Espargaró, A.; Avilés, F.X.; Ventura, S. Detection of transient protein–protein interactions by bimolecular fluorescence complementation: The Abl-SH3 case. Proteomics 2007, 7, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, M.L.; Lamerdin, J.; Owens, S.; Keon, B.H.; Bilter, G.K.; Shang, Z.; Huang, Z.; Yu, H.; Dias, J.; Minami, T.; et al. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat. Chem. Biol. 2006, 2, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Magliery, T.J.; Wilson, C.G.M.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A.D.; Regan, L. Detecting protein-protein interactions with a green fluorescent protein fragment reassembly trap scope and mechanism. J. Am. Chem. Soc. 2005, 127, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Nguyen, H.B.; Pedelacq, F.-D.; Koraïchi, F.; Chaudhary, A.; Ganguly, K.; Lockard, M.A.; Favre, G.; Terwilliger, T.C.; Waldo, G.S. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Pedelacq, J.-D.; Waldo, G.S.; Cabantous, S. High-Throughput Protein-Protein Interaction Assays Using Tripartite Split-GFP Complementation. Methods Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Tripet, B.; Yu, L.; Bautista, D.L.; Wong, W.Y.; Irvin, R.T.; Hodges, R.S. Engineering a de novo-designed coiled-coil heterodimerization domain for the rapid detection, purification and characterization of recombinantly expressed peptides and proteins. Protein Eng. Des. Sel. 1996, 9, 1029–1042. [Google Scholar] [CrossRef]
- Banaszynski, L.A.; Liu, C.W.; Wandless, T.J. Characterization of the FKBP-Rapamycin-FRB Ternary Complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [Google Scholar] [CrossRef]
- Kerppola, T.K. Bimolecular Fluorescence Complementation ( BiFC ) Analysis as a Probe of Protein Interactions in Living Cells. Annu. Rev. Biophys. 2008, 37, 465–487. [Google Scholar] [CrossRef]
- Wehr, M.C.; Rossner, M.J. Split protein biosensor assays in molecular pharmacological studies. Drug Discov. Today 2016, 21, 415–429. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, M.; Tang, R.; Liu, Y.; Lei, C.; Huang, Y.; Nie, Z.; Yao, S. Transpeptidation-Mediated Assembly of Tripartite Split Green Fluorescent Protein for Label-Free Assay of Sortase Activity. Anal. Chem. 2018, 90, 3245–3252. [Google Scholar] [CrossRef] [PubMed]
- Koraïchi, F.; Gence, R.; Bouchenot, C.; Grosjean, S.; Lajoie-Mazenc, I.; Favre, G.; Cabantous, S. High-content tripartite split-GFP cell-based assays to screen for modulators of small GTPase activation. J. Cell Sci. 2018, 131, jcs210419. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Olson, M.F. Purification of TAT-C3 Exoenzyme. Methods Enzymol. 2006, 406, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Afroz, T.; Hock, E.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS- linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017. [Google Scholar] [CrossRef] [PubMed]
- Foglieni, C.; Papin, S.; Salvadè, A.; Afroz, T.; Pinton, S.; Pedrioli, G.; Ulrich, G.; Polymenidou, M.; Paganetti, P. Split GFP technologies to structurally characterize and quantify functional biomolecular interactions of FTD-related proteins. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Lampe, L.; Park, S.; Vangara, B.S.; Waldo, G.S.; Cabantous, S.; Subaran, S.S.; Yang, D.; Lakatta, E.G.; Lin, L. Disulfide Bonds within the C2 Domain of RAGE Play Key Roles in Its Dimerization and Biogenesis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Slingerland, J.; Mari, P.; Nonnekens, J.; Cabantous, S.; Slingerland, J.; Mari, P.; Giglia-Mari, G. In vivo interactions of TTDA mutant proteins within TFIIH. J. Cell Sci. 2013, 126, 3278–3283. [Google Scholar] [CrossRef]
- Finnigan, G.C.; Duvalyan, A.; Liao, E.N.; Sargsyan, A.; Thorner, J.; Pollard, T.D. Detection of protein–protein interactions at the septin collar in Saccharomyces cerevisiae using a tripartite split-GFP system. Mol. Biol. Cell 2016, 27, 2708–2725. [Google Scholar] [CrossRef]
- Chou, W.; Chen, W.; Chu, C.; Dai, C.; Wu, P.; Liu, T.-Y.; Chou, W.; Chen, W.; Chu, C.; Dai, C.; et al. Detection of membrane protein—protein interaction in planta based on dual-intein-coupled tripartite split-GFP association. Plant J. 2018, 94, 426–438. [Google Scholar] [CrossRef]
- Kellermann, S.J.; Rath, A.K.; Rentmeister, A. Tetramolecular Fluorescence Complementation for Detection of Specific RNAs in Vitro. ChemBioChem 2013, 14, 200–204. [Google Scholar] [CrossRef]
- Kellermann, S.J.; Rentmeister, A. A FACS-based screening strategy to assess sequence-specific RNA-binding of Pumilio protein variants in E. coli. Biol. Chem. 2017, 398, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Carlin, K.B.; Cruz-teran, C.A.; Kumar, J.P.; Gomes, C.; Rao, B.M. Combinatorial Pairwise Assembly Efficiently Generates High Affinity Binders and Enables a “Mix-and-Read” Detection Scheme. ACS Synth. Biol. 2016, 5, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate. J. Cachexia Sarcopenia Muscle 2018, 9, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, L.; Wu, H.; Nonell, S.; Dong, Z. Designing a Green Fluorogenic Protease Reporter by Flipping a Beta Strand of GFP for Imaging Apoptosis in Animals. J. Am. Chem. Soc. 2019, 141, 4526–4530. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, S.; Zhang, Z.; Ma, X.; Li, W.; Zhang, X.; Deng, J.; Wei, H.; Li, Z.; Zhang, X.; et al. In vivo imaging of protein–protein and RNA–protein interactions using novel far-red fluorescence complementation systems. Nucleic Acids Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Royant, A.; Lin, M.Z.; Aguilera, T.A.; Lev-Ram, V.; Steinbach, P.A.; Tsien, R.Y. Mammalian Expression of Infrared Fluorescent Proteins Engineered from a Bacterial Phytochrome. Science 2009, 324, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.J.; Lagarias, J.C. Harnessing phytochrome’s glowing potential. Proc. Natl. Acad. Sci. USA 2004, 101, 17334–17339. [Google Scholar] [CrossRef]
- Shcherbakova, D.M.; Baloban, M.; Emelyanov, A.V.; Brenowitz, M.; Guo, P.; Verkhusha, V.V. Bright monomeric near-infrared fluorescent proteins as tags and biosensors for multiscale imaging. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Tchekanda, E.; Sivanesan, D. An infrared reporter to detect spatiotemporal dynamics of protein- protein interactions. Nat. Methods 2014, 11. [Google Scholar] [CrossRef]
- To, T.; Zhang, Q.; Shu, X. Structure-guided design of a reversible fluorogenic reporter of protein-protein interactions. Protein Sci. 2016, 25, 748–753. [Google Scholar] [CrossRef]
- Köker, T.; Fernandez, A.; Pinaud, F. Characterization of Split Fluorescent Protein Variants and Quantitative Analyses of Their Self- Assembly Process. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Both, J.; Do, K.; Boxer, S.G. Mechanism and bottlenecks in strand photodissociation of split green fluorescent proteins (GFPs). Proc. Natl. Acad. Sci. USA 2017, E2146–E2155. [Google Scholar] [CrossRef] [PubMed]
- Kent, K.P.; Boxer, S.G. Light-Activated Reassembly of Split Green Fluorescent Protein. J. Am. Chem. Soc. 2011, 133, 4046–4052. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Bystroff, C. Complementation and Reconstitution of Fluorescence from Circularly Permuted and Truncated Green Fluorescent Protein. Biochemistry 2009, 48, 929–940. [Google Scholar] [CrossRef]
- Lundqvist, M.; Thalén, N.; Volk, A.; Hansen, H.G.; von Otter, E.; Nygren, P.-Å.; Uhlen, M.; Rockberg, J. Chromophore pre-maturation for improved speed and sensitivity of split-GFP monitoring of protein secretion. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Dong, X.; Jiang, J.; Yang, Y.; Yang, J.; Lu, Y. Specific cell surface labeling of GPCRs using split GFP. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kirchhofer, A.; Helma, J.; Schmidthals, K.; Frauer, C.; Cui, S.; Karcher, A.; Pellis, M.; Muyldermans, S.; Casas-delucchi, C.S.; Cardoso, M.C.; et al. Modulation of protein properties in living cells using nanobodies. Nat. Struct. Mol. Biol. 2009, 17, 133–138. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedelacq, J.-D.; Cabantous, S. Development and Applications of Superfolder and Split Fluorescent Protein Detection Systems in Biology. Int. J. Mol. Sci. 2019, 20, 3479. https://doi.org/10.3390/ijms20143479
Pedelacq J-D, Cabantous S. Development and Applications of Superfolder and Split Fluorescent Protein Detection Systems in Biology. International Journal of Molecular Sciences. 2019; 20(14):3479. https://doi.org/10.3390/ijms20143479
Chicago/Turabian StylePedelacq, Jean-Denis, and Stéphanie Cabantous. 2019. "Development and Applications of Superfolder and Split Fluorescent Protein Detection Systems in Biology" International Journal of Molecular Sciences 20, no. 14: 3479. https://doi.org/10.3390/ijms20143479
APA StylePedelacq, J.-D., & Cabantous, S. (2019). Development and Applications of Superfolder and Split Fluorescent Protein Detection Systems in Biology. International Journal of Molecular Sciences, 20(14), 3479. https://doi.org/10.3390/ijms20143479