Defining Bronchial Asthma with Phosphoinositide 3-Kinase Delta Activation: Towards Endotype-Driven Management


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Introduction to Class I PI3K and Its Isoforms
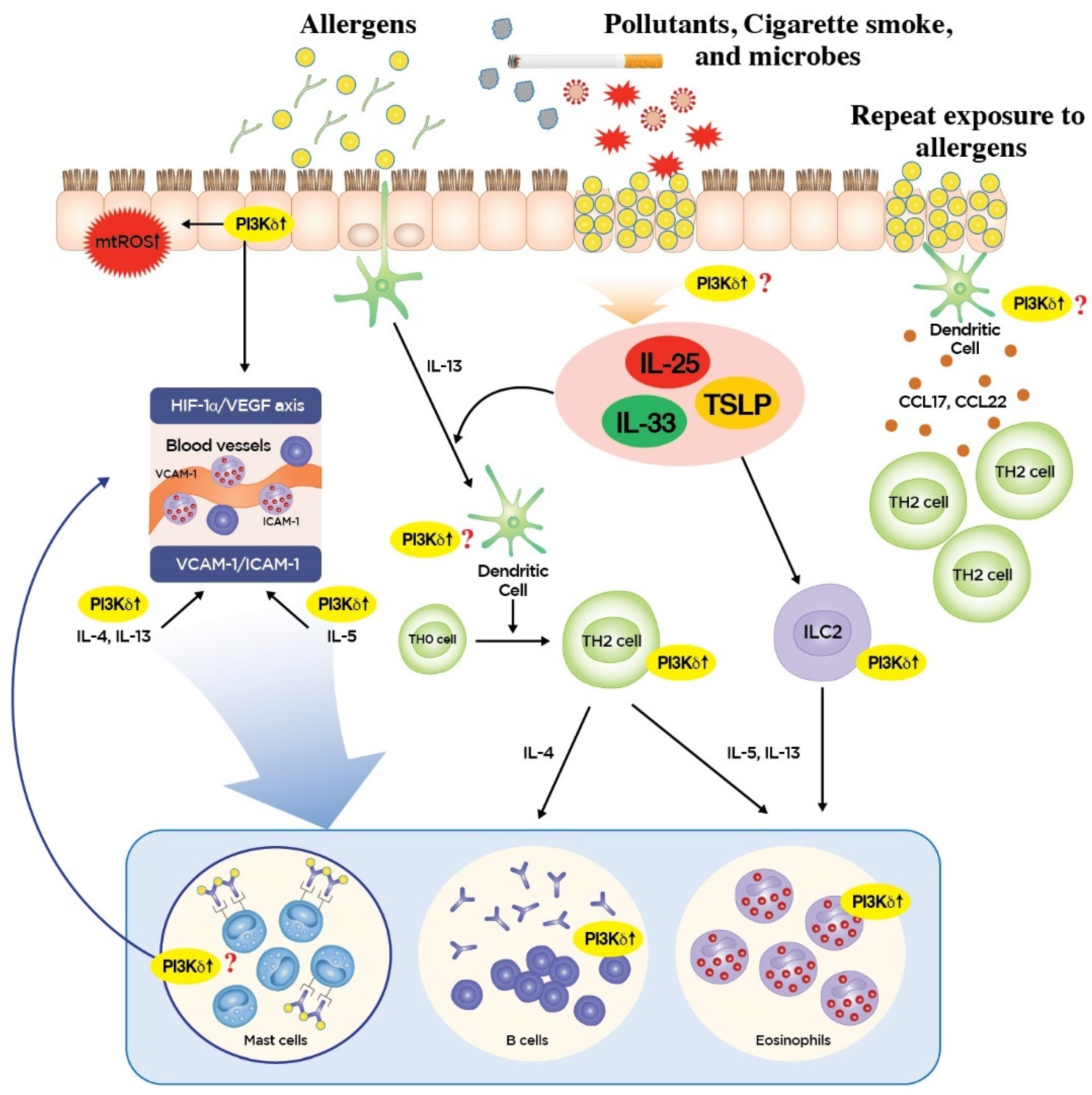
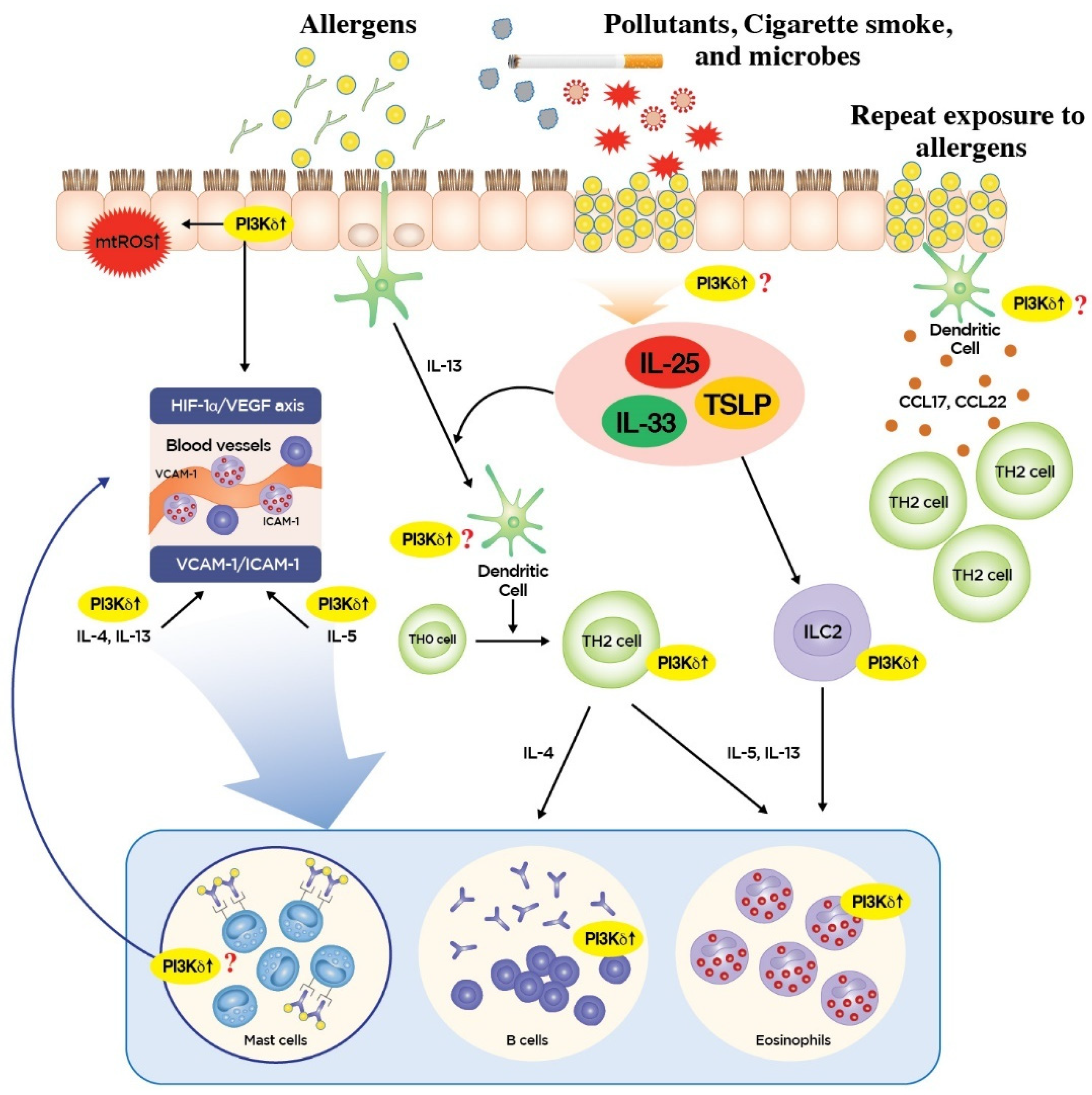
3. Roles of PI3K-δ in Type 2 Inflammation
4. Roles of PI3K-δ in Non-Type 2 Inflammation
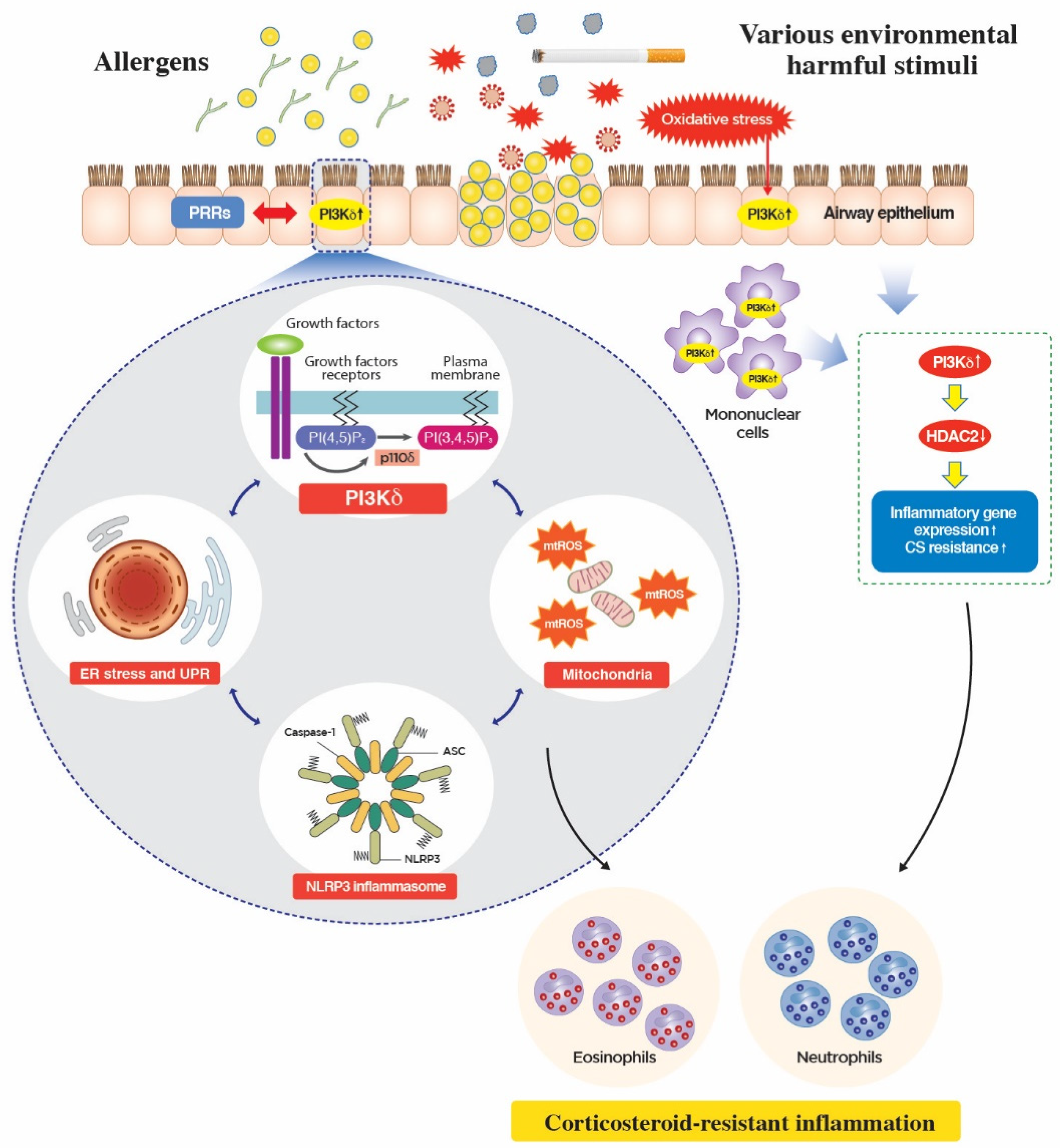
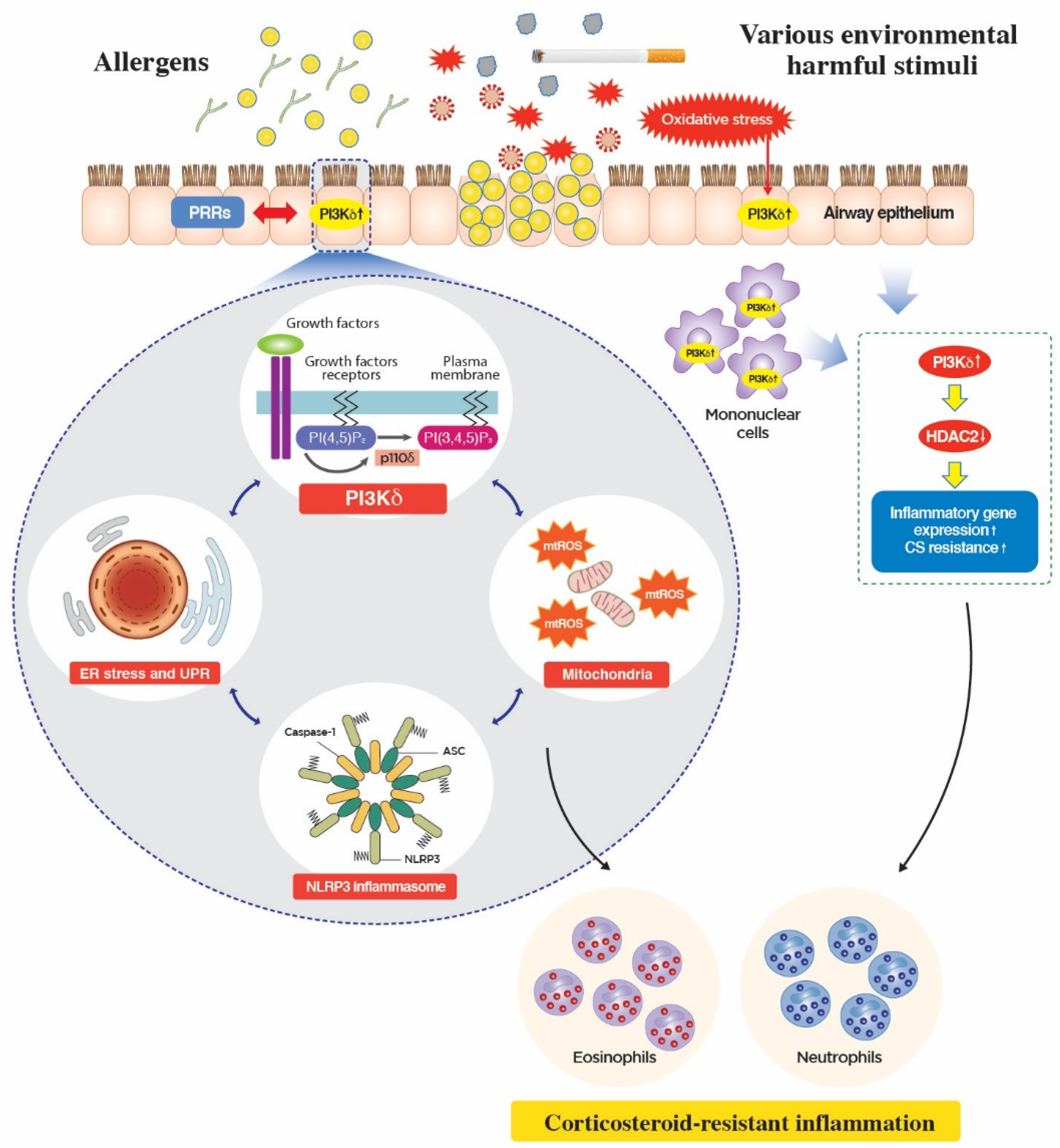
5. Novel Mechanism of Action of PI3K-δ in CS-Resistant Lung Inflammation
6. PI3K-δ Inhibitors in Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ray, A.; Raundhal, M.; Oriss, T.B.; Ray, P.; Wenzel, S.E. Current concepts of severe asthma. J. Clin. Investig. 2016, 126, 2394–2403. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Kim, S.R.; Cho, S.H.; Lee, Y.C. A Novel Insight on Endotyping Heterogeneous Severe Asthma Based on Endoplasmic Reticulum Stress: Beyond the “Type 2/Non-Type 2 Dichotomy”. Int. J. Mol. Sci. 2019, 20, 713. [Google Scholar] [CrossRef] [PubMed]
- Carr, T.F.; Zeki, A.A.; Kraft, M. Eosinophilic and Noneosinophilic Asthma. Am. J. Respir. Crit. Care Med. 2018, 197, 22–37. [Google Scholar] [CrossRef] [PubMed]
- McGregor, M.C.; Krings, J.G.; Nair, P.; Castro, M. Role of Biologics in Asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Krings, J.G.; McGregor, M.C.; Bacharier, L.B.; Castro, M. Biologics for Severe Asthma: Treatment-Specific Effects Are Important in Choosing a Specific Agent. J. Allergy Clin. Immunol. Pract. 2019, 7, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2, 807–808. [Google Scholar]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef] [Green Version]
- Bi, L.; Okabe, I.; Bernard, D.J.; Wynshaw-Boris, A.; Nussbaum, R.L. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J. Biol. Chem. 1999, 274, 10963–10968. [Google Scholar] [CrossRef] [PubMed]
- Bi, L.; Okabe, I.; Bernard, D.J.; Nussbaum, R.L. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm. Genome 2002, 13, 169–172. [Google Scholar] [PubMed]
- Barnes, P.J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2013, 131, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Stokes, C.A.; Condliffe, A.M. Phosphoinositide 3-kinase delta (PI3Kdelta) in respiratory disease. Biochem. Soc. Trans. 2018, 46, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Jeong, J.S.; Kim, S.R.; Cho, S.H.; Kolliputi, N.; Ko, Y.H.; Lee, K.B.; Park, S.C.; Park, H.J.; Lee, Y.C. Phosphoinositide 3-kinase-delta regulates fungus-induced allergic lung inflammation through endoplasmic reticulum stress. Thorax 2016, 71, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Lee, K.B.; Kim, S.R.; Kim, D.I.; Park, H.J.; Lee, H.K.; Kim, H.J.; Cho, S.H.; Kolliputi, N.; Kim, S.H.; et al. Airway epithelial phosphoinositide 3-kinase-delta contributes to the modulation of fungi-induced innate immune response. Thorax 2018, 73, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Coulter, T.I.; Chandra, A.; Bacon, C.M.; Babar, J.; Curtis, J.; Screaton, N.; Goodlad, J.R.; Farmer, G.; Steele, C.L.; Leahy, T.R.; et al. Clinical spectrum and features of activated phosphoinositide 3-kinase delta syndrome: A large patient cohort study. J. Allergy Clin. Immunol. 2017, 139, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Angulo, I.; Vadas, O.; Garcon, F.; Banham-Hall, E.; Plagnol, V.; Leahy, T.R.; Baxendale, H.; Coulter, T.; Curtis, J.; Wu, C.; et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science 2013, 342, 866–871. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H. The immunology of asthma. Nat. Immunol. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Doherty, T.A.; Khorram, N.; Lund, S.; Mehta, A.K.; Croft, M.; Broide, D.H. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J. Allergy Clin. Immunol. 2013, 132, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Verma, M.; Michalec, L.; Liu, W.; Sripada, A.; Rollins, D.; Good, J.; Ito, Y.; Chu, H.; Gorska, M.M.; et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2018, 141, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Millien, V.O.; Lu, W.; Shaw, J.; Yuan, X.; Mak, G.; Roberts, L.; Song, L.Z.; Knight, J.M.; Creighton, C.J.; Luong, A.; et al. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science 2013, 341, 792–796. [Google Scholar] [CrossRef] [PubMed]
- Salmond, R.J.; Mirchandani, A.S.; Besnard, A.G.; Bain, C.C.; Thomson, N.C.; Liew, F.Y. IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J. Allergy Clin. Immunol. 2012, 130, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Kim, D.I.; Kim, S.H.; Lee, H.; Lee, K.S.; Cho, S.H.; Lee, Y.C. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014, 5, e1498. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, K.S.; Park, H.S.; Park, S.J.; Min, K.H.; Moon, H.; Puri, K.D.; Lee, Y.C. HIF-1alpha inhibition ameliorates an allergic airway disease via VEGF suppression in bronchial epithelium. Eur. J. Immunol. 2010, 40, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Min, K.H.; Lee, K.Y.; Choe, Y.H.; Park, S.Y.; Chai, O.H.; Zhang, X.; Song, C.H.; et al. Mast cells can mediate vascular permeability through regulation of the PI3K-HIF-1alpha-VEGF axis. Am. J. Respir. Crit. Care Med. 2008, 178, 787–797. [Google Scholar] [CrossRef]
- Aksoy, E.; Saveanu, L.; Manoury, B. The Isoform Selective Roles of PI3Ks in Dendritic Cell Biology and Function. Front. Immunol. 2018, 9, 2574. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, E.; Taboubi, S.; Torres, D.; Delbauve, S.; Hachani, A.; Whitehead, M.A.; Pearce, W.P.; Berenjeno, I.M.; Nock, G.; Filloux, A.; et al. The p110delta isoform of the kinase PI(3)K controls the subcellular compartmentalization of TLR4 signaling and protects from endotoxic shock. Nat. Immunol. 2012, 13, 1045–1054. [Google Scholar] [CrossRef]
- Horak, F.; Puri, K.D.; Steiner, B.H.; Holes, L.; Xing, G.; Zieglmayer, P.; Zieglmayer, R.; Lemell, P.; Yu, A. Randomized phase 1 study of the phosphatidylinositol 3-kinase delta inhibitor idelalisib in patients with allergic rhinitis. J. Allergy Clin. Immunol. 2016, 137, 1733–1741. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Turner, M.; Gold, M.R. PI3K Signaling in B Cell and T Cell Biology. Front. Immunol. 2014, 5, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Snapper, S.B.; Yballe, C.M.; Davidson, L.; Yu, J.Y.; Alt, F.W.; Cantley, L.C. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science 1999, 283, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Patton, D.T.; Bilancio, A.; Garcon, F.; Rowan, W.C.; Vanhaesebroeck, B. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J. Immunol. 2006, 177, 5122–5128. [Google Scholar] [CrossRef] [PubMed]
- Jarmin, S.J.; David, R.; Ma, L.; Chai, J.G.; Dewchand, H.; Takesono, A.; Ridley, A.J.; Okkenhaug, K.; Marelli-Berg, F.M. T cell receptor-induced phosphoinositide-3-kinase p110delta activity is required for T cell localization to antigenic tissue in mice. J. Clin. Investig. 2008, 118, 1154–1164. [Google Scholar] [PubMed]
- Soond, D.R.; Bjorgo, E.; Moltu, K.; Dale, V.Q.; Patton, D.T.; Torgersen, K.M.; Galleway, F.; Twomey, B.; Clark, J.; Gaston, J.S.; et al. PI3K p110delta regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood 2010, 115, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.L.; Schwartz, M.D.; Jameson, S.C.; Shimizu, Y. Selective regulation of CD8 effector T cell migration by the p110 gamma isoform of phosphatidylinositol 3-kinase. J. Immunol. 2008, 180, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Bilancio, A.; Okkenhaug, K.; Camps, M.; Emery, J.L.; Ruckle, T.; Rommel, C.; Vanhaesebroeck, B. Key role of the p110delta isoform of PI3K in B-cell antigen and IL-4 receptor signaling: Comparative analysis of genetic and pharmacologic interference with p110delta function in B cells. Blood 2006, 107, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Omori, S.A.; Cato, M.H.; Anzelon-Mills, A.; Puri, K.D.; Shapiro-Shelef, M.; Calame, K.; Rickert, R.C. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity 2006, 25, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.; Bardi, G.; Bell, S.E.; Chantry, D.; Downes, C.P.; Gray, A.; Humphries, L.A.; Rawlings, D.; Reynolds, H.; Vigorito, E.; et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J. Exp. Med. 2002, 196, 753–763. [Google Scholar] [CrossRef]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.T.; Okkenhaug, K.; Nashed, B.F.; Puri, K.D.; Knight, Z.A.; Shokat, K.M.; Vanhaesebroeck, B.; Marshall, A.J. Genetic or pharmaceutical blockade of p110delta phosphoinositide 3-kinase enhances IgE production. J. Allergy Clin. Immunol. 2008, 122, 811–819. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, H.K.; Hayflick, J.S.; Lee, Y.C.; Puri, K.D. Inhibition of phosphoinositide 3-kinase delta attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB J. 2006, 20, 455–465. [Google Scholar] [CrossRef]
- Rosenberg, H.F.; Phipps, S.; Foster, P.S. Eosinophil trafficking in allergy and asthma. J. Allergy Clin. Immunol. 2007, 119, 1303–1310. [Google Scholar] [CrossRef]
- Kang, B.N.; Ha, S.G.; Ge, X.N.; Reza Hosseinkhani, M.; Bahaie, N.S.; Greenberg, Y.; Blumenthal, M.N.; Puri, K.D.; Rao, S.P.; Sriramarao, P. The p110delta subunit of PI3K regulates bone marrow-derived eosinophil trafficking and airway eosinophilia in allergen-challenged mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L1179–L1191. [Google Scholar] [CrossRef] [PubMed]
- Cherry, W.B.; Yoon, J.; Bartemes, K.R.; Iijima, K.; Kita, H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J. Allergy Clin. Immunol. 2008, 121, 1484–1490. [Google Scholar] [CrossRef] [Green Version]
- Na, H.J.; Hudson, S.A.; Bochner, B.S. IL-33 enhances Siglec-8 mediated apoptosis of human eosinophils. Cytokine 2012, 57, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.K.; Leung, K.M.; Qiu, H.N.; Chow, J.Y.; Choi, A.O.; Lam, C.W. Activation of eosinophils interacting with dermal fibroblasts by pruritogenic cytokine IL-31 and alarmin IL-33: Implications in atopic dermatitis. PLoS ONE 2012, 7, e29815. [Google Scholar] [CrossRef] [PubMed]
- Kvarnhammar, A.M.; Cardell, L.O. Pattern-recognition receptors in human eosinophils. Immunology 2012, 136, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dil, N.; Marshall, A.J. Role of phosphoinositide 3-kinase p110 delta in TLR4- and TLR9-mediated B cell cytokine production and differentiation. Mol. Immunol. 2009, 46, 1970–1978. [Google Scholar] [CrossRef] [PubMed]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; Dupont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A.; et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef]
- Al-Ramli, W.; Prefontaine, D.; Chouiali, F.; Martin, J.G.; Olivenstein, R.; Lemiere, C.; Hamid, Q. T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J. Allergy Clin. Immunol. 2009, 123, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Ricciardolo, F.L.M.; Sorbello, V.; Folino, A.; Gallo, F.; Massaglia, G.M.; Favata, G.; Conticello, S.; Vallese, D.; Gani, F.; Malerba, M.; et al. Identification of IL-17F/frequent exacerbator endotype in asthma. J. Allergy Clin. Immunol. 2017, 140, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Raundhal, M.; Morse, C.; Khare, A.; Oriss, T.B.; Milosevic, J.; Trudeau, J.; Huff, R.; Pilewski, J.; Holguin, F.; Kolls, J.; et al. High IFN-gamma and low SLPI mark severe asthma in mice and humans. J. Clin. Investig. 2015, 125, 3037–3050. [Google Scholar] [CrossRef] [PubMed]
- Duvall, M.G.; Barnig, C.; Cernadas, M.; Ricklefs, I.; Krishnamoorthy, N.; Grossman, N.L.; Bhakta, N.R.; Fahy, J.V.; Bleecker, E.R.; Castro, M.; et al. Natural killer cell-mediated inflammation resolution is disabled in severe asthma. Sci. Immunol. 2017, 2, eaam5446. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.L.; Trudeau, J.B.; Scheller, E.V.; Mandalapu, S.; Elloso, M.M.; Kolls, J.K.; Wenzel, S.E.; Alcorn, J.F. The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunol. 2014, 7, 1186–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modena, B.D.; Tedrow, J.R.; Milosevic, J.; Bleecker, E.R.; Meyers, D.A.; Wu, W.; Bar-Joseph, Z.; Erzurum, S.C.; Gaston, B.M.; Busse, W.W.; et al. Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am. J. Respir. Crit. Care Med. 2014, 190, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.S.; Pavlidis, S.; Loza, M.; Baribaud, F.; Rowe, A.; Pandis, I.; Sousa, A.; Corfield, J.; Djukanovic, R.; Lutter, R.; et al. T-helper cell type 2 (Th2) and non-Th2 molecular phenotypes of asthma using sputum transcriptomics in U-BIOPRED. Eur. Respir. J. 2017, 49, 1602135. [Google Scholar] [CrossRef] [PubMed]
- Kwak, Y.G.; Song, C.H.; Yi, H.K.; Hwang, P.H.; Kim, J.S.; Lee, K.S.; Lee, Y.C. Involvement of PTEN in airway hyperresponsiveness and inflammation in bronchial asthma. J. Clin. Investig. 2003, 111, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Ali, K.; Bilancio, A.; Thomas, M.; Pearce, W.; Gilfillan, A.M.; Tkaczyk, C.; Kuehn, N.; Gray, A.; Giddings, J.; Peskett, E.; et al. Essential role for the p110delta phosphoinositide 3-kinase in the allergic response. Nature 2004, 431, 1007–1011. [Google Scholar] [CrossRef]
- Nashed, B.F.; Zhang, T.; Al-Alwan, M.; Srinivasan, G.; Halayko, A.J.; Okkenhaug, K.; Vanhaesebroeck, B.; Hayglass, K.T.; Marshall, A.J. Role of the phosphoinositide 3-kinase p110delta in generation of type 2 cytokine responses and allergic airway inflammation. Eur. J. Immunol. 2007, 37, 416–424. [Google Scholar] [CrossRef]
- Myou, S.; Leff, A.R.; Myo, S.; Boetticher, E.; Tong, J.; Meliton, A.Y.; Liu, J.; Munoz, N.M.; Zhu, X. Blockade of Inflammation and Airway Hyperresponsiveness in Immune-sensitized Mice by Dominant-Negative Phosphoinositide 3-Kinase–TAT. J. Exp. Med. 2003, 198, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Patton, D.T.; Garden, O.A.; Pearce, W.P.; Clough, L.E.; Monk, C.R.; Leung, E.; Rowan, W.C.; Sancho, S.; Walker, L.S.; Vanhaesebroeck, B.; et al. Cutting edge: The phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. 2006, 177, 6598–6602. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, E.C.; Kobayashi, T.; Russo, S.M.; Sheikh, S.Z.; Gipson, G.R.; Kennedy, S.T.; Uno, J.K.; Mishima, Y.; Borst, L.B.; Liu, B.; et al. Innate PI3K p110delta regulates Th1/Th17 development and microbiota-dependent colitis. J. Immunol. 2014, 192, 3958–3968. [Google Scholar] [CrossRef] [PubMed]
- Way, E.E.; Trevejo-Nunez, G.; Kane, L.P.; Steiner, B.H.; Puri, K.D.; Kolls, J.K.; Chen, K. Dose-Dependent Suppression of Cytokine production from T cells by a Novel Phosphoinositide 3-Kinase Delta Inhibitor. Sci. Rep. 2016, 6, 30384. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Lee, K.S.; Kim, S.R.; Min, K.H.; Moon, H.; Lee, M.H.; Chung, C.R.; Han, H.J.; Puri, K.D.; Lee, Y.C. Phosphoinositide 3-kinase delta inhibitor suppresses interleukin-17 expression in a murine asthma model. Eur. Respir. J. 2010, 36, 1448–1459. [Google Scholar] [CrossRef] [PubMed]
- Roller, A.; Perino, A.; Dapavo, P.; Soro, E.; Okkenhaug, K.; Hirsch, E.; Ji, H. Blockade of phosphatidylinositol 3-kinase PI3Kdelta or PI3Kgamma reduces IL-17 and ameliorates imiquimod-induced psoriasis-like dermatitis. J. Immunol. 2012, 189, 4612–4620. [Google Scholar] [CrossRef]
- Low, P.C.; Manzanero, S.; Mohannak, N.; Narayana, V.K.; Nguyen, T.H.; Kvaskoff, D.; Brennan, F.H.; Ruitenberg, M.J.; Gelderblom, M.; Magnus, T.; et al. PI3Kdelta inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model. Nat. Commun. 2014, 5, 3450. [Google Scholar] [CrossRef]
- Low, P.C.; Misaki, R.; Schroder, K.; Stanley, A.C.; Sweet, M.J.; Teasdale, R.D.; Vanhaesebroeck, B.; Meunier, F.A.; Taguchi, T.; Stow, J.L. Phosphoinositide 3-kinase delta regulates membrane fission of Golgi carriers for selective cytokine secretion. J. Cell Biol. 2010, 190, 1053–1065. [Google Scholar] [CrossRef]
- Sadhu, C.; Masinovsky, B.; Dick, K.; Sowell, C.G.; Staunton, D.E. Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J. Immunol. 2003, 170, 2647–2654. [Google Scholar] [CrossRef]
- Puri, K.D.; Doggett, T.A.; Douangpanya, J.; Hou, Y.; Tino, W.T.; Wilson, T.; Graf, T.; Clayton, E.; Turner, M.; Hayflick, J.S.; et al. Mechanisms and implications of phosphoinositide 3-kinase delta in promoting neutrophil trafficking into inflamed tissue. Blood 2004, 103, 3448–3456. [Google Scholar] [CrossRef]
- Sadhu, C.; Dick, K.; Tino, W.T.; Staunton, D.E. Selective role of PI3K delta in neutrophil inflammatory responses. Biochem. Biophys. Res. Commun. 2003, 308, 764–769. [Google Scholar] [CrossRef]
- Ito, K.; Ito, M.; Elliott, W.M.; Cosio, B.; Caramori, G.; Kon, O.M.; Barczyk, A.; Hayashi, S.; Adcock, I.M.; Hogg, J.C.; et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 2005, 352, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, G.J.; Ten Hacken, N.H.; Hoffmann, R.F.; van Oosterhout, A.J.; Heijink, I.H. Interleukin-17A induces glucocorticoid insensitivity in human bronchial epithelial cells. Eur. Respir. J. 2012, 39, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Marwick, J.A.; Caramori, G.; Casolari, P.; Mazzoni, F.; Kirkham, P.A.; Adcock, I.M.; Chung, K.F.; Papi, A. A role for phosphoinositol 3-kinase delta in the impairment of glucocorticoid responsiveness in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2010, 125, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bleecker, E.; Moore, W.; Busse, W.W.; Castro, M.; Chung, K.F.; Calhoun, W.J.; Erzurum, S.; Gaston, B.; Israel, E.; et al. Unsupervised phenotyping of Severe Asthma Research Program participants using expanded lung data. J. Allergy Clin. Immunol. 2014, 133, 1280–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F., Jr.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 2011, 127, 355–360. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, S.R.; Lee, Y.C. Can Controlling Endoplasmic Reticulum Dysfunction Treat Allergic Inflammation in Severe Asthma with Fungal Sensitization? Allergy Asthma Immunol. Res. 2018, 10, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.S.; Kim, S.R.; Cho, S.H.; Lee, Y.C. Endoplasmic Reticulum Stress and Allergic Diseases. Curr. Allergy Asthma Rep. 2017, 17, 82. [Google Scholar] [CrossRef]
- Park, S.W.; Zhou, Y.; Lee, J.; Lu, A.; Sun, C.; Chung, J.; Ueki, K.; Ozcan, U. The regulatory subunits of PI3K, p85alpha and p85beta, interact with XBP-1 and increase its nuclear translocation. Nat. Med. 2010, 16, 429–437. [Google Scholar] [CrossRef]
- Zhang, W.; Neo, S.P.; Gunaratne, J.; Poulsen, A.; Boping, L.; Ong, E.H.; Sangthongpitag, K.; Pendharkar, V.; Hill, J.; Cohen, S.M. Feedback regulation on PTEN/AKT pathway by the ER stress kinase PERK mediated by interaction with the Vault complex. Cell. Signal. 2015, 27, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef] [PubMed]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nunez, G.; He, Y.; Yin, X.M.; O’Riordan, M.X. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Lee, M.H.; Jin, S.M.; Jin, G.Y.; Yoo, W.H.; Lee, Y.C. Hydrogen peroxide induces vascular permeability via regulation of vascular endothelial growth factor. Am. J. Respir. Cell Mol. Biol. 2006, 35, 190–197. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Sternberg, P.; Cai, J. Essential roles of the PI3 kinase/Akt pathway in regulating Nrf2-dependent antioxidant functions in the RPE. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1671–1678. [Google Scholar] [CrossRef]
- Ciencewicki, J.; Trivedi, S.; Kleeberger, S.R. Oxidants and the pathogenesis of lung diseases. J. Allergy Clin. Immunol. 2008, 122, 456–468. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, Z.Z.; Wang, W. Inhibition of endoplasmic reticulum stress alleviates cigarette smoke-induced airway inflammation and emphysema. Oncotarget 2017, 8, 77685–77695. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, Y.C. Endoplasmic reticulum stress and the related signaling networks in severe asthma. Allergy Asthma Immunol. Res. 2015, 7, 106–117. [Google Scholar] [CrossRef]
- Kim, R.Y.; Pinkerton, J.W.; Essilfie, A.T.; Robertson, A.A.B.; Baines, K.J.; Brown, A.C.; Mayall, J.R.; Ali, M.K.; Starkey, M.R.; Hansbro, N.G.; et al. Role for NLRP3 Inflammasome-mediated, IL-1beta-Dependent Responses in Severe, Steroid-Resistant Asthma. Am. J. Respir. Crit. Care Med. 2017, 196, 283–297. [Google Scholar] [CrossRef]
- Simpson, J.L.; Phipps, S.; Baines, K.J.; Oreo, K.M.; Gunawardhana, L.; Gibson, P.G. Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur. Respir. J. 2014, 43, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Kinases as Novel Therapeutic Targets in Asthma and Chronic Obstructive Pulmonary Disease. Pharmacol. Rev. 2016, 68, 788–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Down, K.; Amour, A.; Baldwin, I.R.; Cooper, A.W.; Deakin, A.M.; Felton, L.M.; Guntrip, S.B.; Hardy, C.; Harrison, Z.A.; Jones, K.L.; et al. Optimization of Novel Indazoles as Highly Potent and Selective Inhibitors of Phosphoinositide 3-Kinase δ for the Treatment of Respiratory Disease. J. Med. Chem. 2015, 58, 7381–7399. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef]
- Stark, A.K.; Sriskantharajah, S.; Hessel, E.M.; Okkenhaug, K. PI3K inhibitors in inflammation, autoimmunity and cancer. Curr. Opin. Pharmacol. 2015, 23, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, J.S.; Kim, J.S.; Kim, S.R.; Lee, Y.C. Defining Bronchial Asthma with Phosphoinositide 3-Kinase Delta Activation: Towards Endotype-Driven Management. Int. J. Mol. Sci. 2019, 20, 3525. https://doi.org/10.3390/ijms20143525
Jeong JS, Kim JS, Kim SR, Lee YC. Defining Bronchial Asthma with Phosphoinositide 3-Kinase Delta Activation: Towards Endotype-Driven Management. International Journal of Molecular Sciences. 2019; 20(14):3525. https://doi.org/10.3390/ijms20143525
Chicago/Turabian StyleJeong, Jae Seok, Jong Seung Kim, So Ri Kim, and Yong Chul Lee. 2019. "Defining Bronchial Asthma with Phosphoinositide 3-Kinase Delta Activation: Towards Endotype-Driven Management" International Journal of Molecular Sciences 20, no. 14: 3525. https://doi.org/10.3390/ijms20143525
APA StyleJeong, J. S., Kim, J. S., Kim, S. R., & Lee, Y. C. (2019). Defining Bronchial Asthma with Phosphoinositide 3-Kinase Delta Activation: Towards Endotype-Driven Management. International Journal of Molecular Sciences, 20(14), 3525. https://doi.org/10.3390/ijms20143525