Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy



Abstract
1. Introduction
1.1. Fatty Acid Metabolism
1.2. Acyl-CoA-Synthetases
 Acyl-AMP + PPi
Acyl-AMP + PPi
 Acyl-CoA + AMP
Acyl-CoA + AMP
2. ACSLs in Cancer
2.1. Colorectal Cancer
2.2. Breast Cancer
2.3. Prostate Cancer
2.4. Melanoma
2.5. Liver Cancer
2.6. Lung Cancer
2.7. Soft Tissue Cancers
2.8. Blood Cancers
3. Pharmacological Targeting of ACSLs
4. ACSLs as Therapeutic Targets in Cancer
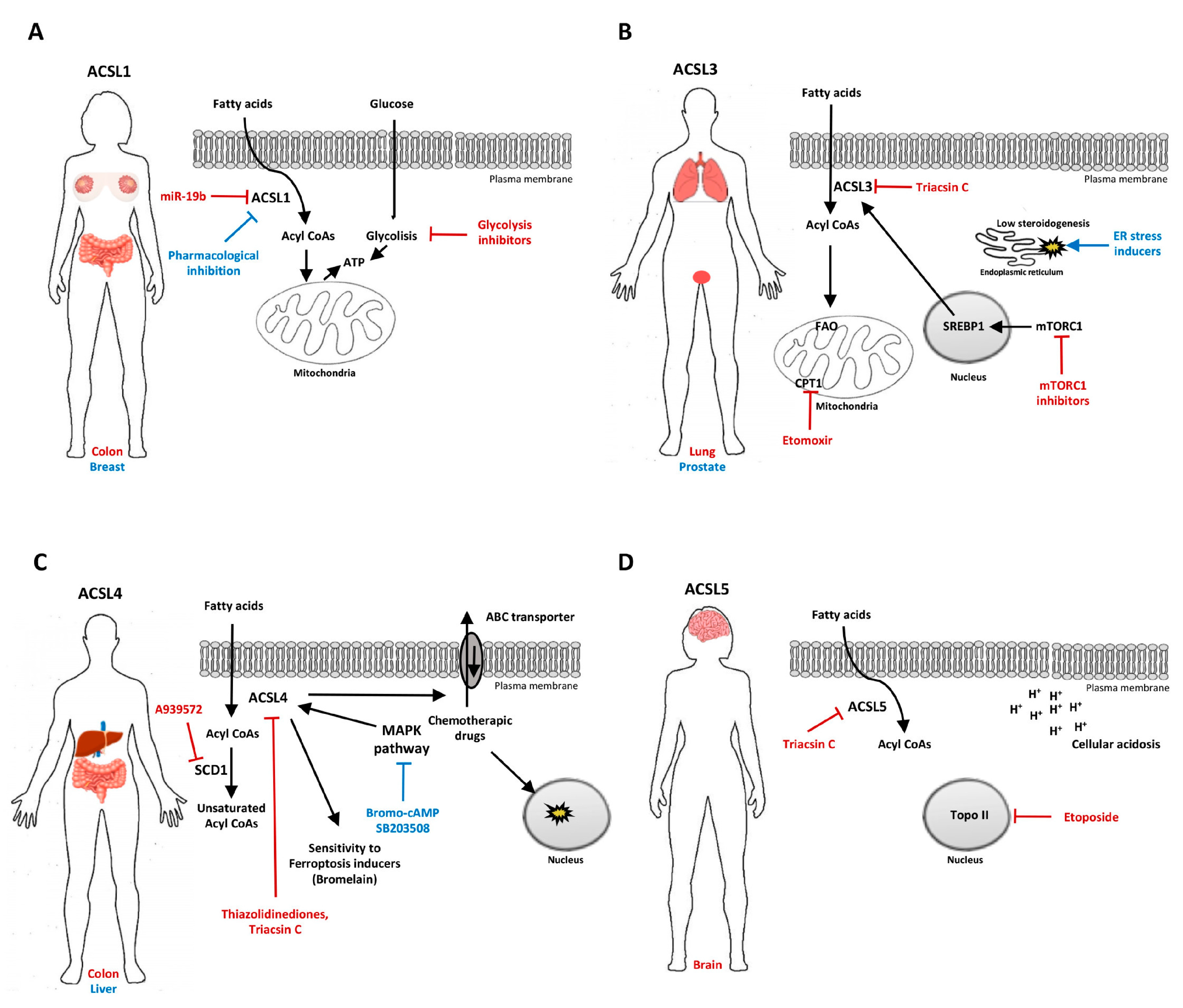
4.1. ACSL1
4.2. ACSL3
4.3. ACSL4
4.4. ACSL5
4.5. ACSL6
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular carcinoma |
| ER | Estrogen receptor |
| CRC | Colorectal Carcinoma |
| ACSL1 | Acyl CoA long chain synthetase 1 |
| ACSL3 | Acyl CoA long chain synthetase 3 |
| ACSL4 | Acyl CoA long chain synthetase 4 |
| ACSL5 | Acyl CoA long chain synthetase 5 |
| ACSL6 | Acyl CoA long chain synthetase 6 |
| EMT | Epithelial to Mesenchymal transition |
| SCD1 | Stearoyl-CoA Desaturase 1 |
| CRPC | Castration-resistant prostate cancer |
| BC | Breast cancer |
| FAO | Fatty Acid Oxidation |
| NSCLC | Non-small cell lung cancer |
References
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell. Metab. 2013, 18, 153–161. [Google Scholar] [PubMed]
- Ellis, J.M.; Li, L.O.; Wu, P.C.; Koves, T.R.; Ilkayeva, O.; Stevens, R.D.; Watkins, S.M.; Muoio, D.M.; Coleman, R.A. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell. Metab. 2010, 12, 53–64. [Google Scholar] [PubMed]
- Grevengoed, T.J.; Klett, E.L.; Coleman, R.A. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014, 34, 1–30. [Google Scholar] [PubMed]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.M.; Lu, G.D. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [PubMed]
- Khalid, A.; Siddiqui, A.J.; Huang, J.H.; Shamsi, T.; Musharraf, S.G. Alteration of serum free fatty acids are indicators for progression of pre-leukaemia diseases to leukaemia. Sci. Rep. 2018, 8, 14883. [Google Scholar] [PubMed]
- Ren, J.; Zhang, D.; Liu, Y.; Zhang, R.; Fang, H.; Guo, S.; Zhou, D.; Zhang, M.; Xu, Y.; Qiu, L.; et al. Simultaneous quantification of serum nonesterified and esterified fatty acids as potential biomarkers to differentiate benign lung diseases from lung cancer. Sci. Rep. 2016, 6, 34201. [Google Scholar] [PubMed]
- Zhang, Y.; He, C.; Qiu, L.; Wang, Y.; Zhang, L.; Qin, X.; Liu, Y.; Zhang, D.; Li, Z. Serum unsaturated free fatty acids: Potential biomarkers for early detection and disease progression monitoring of non-small cell lung cancer. J. Cancer 2014, 5, 706–714. [Google Scholar]
- Shaikh, S.; Channa, N.A.; Talpur, F.N.; Younis, M.; Tabassum, N. Radiotherapy improves serum fatty acids and lipid profile in breast cancer. Lipids Health Dis. 2017, 16, 92. [Google Scholar]
- Zhang, Y.; He, C.; Qiu, L.; Wang, Y.; Qin, X.; Liu, Y.; Li, Z. Serum unsaturated free fatty acids: A potential biomarker panel for early-stage detection of colorectal cancer. J. Cancer 2016, 7, 477–483. [Google Scholar]
- Iemoto, T.; Nishiumi, S.; Kobayashi, T.; Fujigaki, S.; Hamaguchi, T.; Kato, K.; Shoji, H.; Matsumura, Y.; Honda, K.; Yoshida, M. Serum level of octanoic acid predicts the efficacy of chemotherapy for colorectal cancer. Oncol. Lett. 2019, 17, 831–842. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [PubMed]
- Balaban, S.; Nassar, Z.D.; Zhang, A.Y.; Hosseini-Beheshti, E.; Centenera, M.M.; Schreuder, M.; Lin, H.M.; Aishah, A.; Varney, B.; Liu-Fu, F.; et al. Extracellular fatty acids are the major contributor to lipid synthesis in prostate cancer. Mol. Cancer Res. 2019, 17, 949–962. [Google Scholar] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [PubMed]
- Porstmann, T.; Griffiths, B.; Chung, Y.L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. Pkb/akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of srebp. Oncogene 2005, 24, 6465–6481. [Google Scholar]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.P.; Wang, B.; Cohen, A.; et al. Scd1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 2012, 7, e33823. [Google Scholar]
- Peck, B.; Schulze, A. Lipid desaturation—The next step in targeting lipogenesis in cancer? FEBS J. 2016, 283, 2767–2778. [Google Scholar]
- Vriens, K.; Christen, S.; Parik, S.; Broekaert, D.; Yoshinaga, K.; Talebi, A.; Dehairs, J.; Escalona-Noguero, C.; Schmieder, R.; Cornfield, T.; et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 2019, 566, 403–406. [Google Scholar]
- Padanad, M.S.; Konstantinidou, G.; Venkateswaran, N.; Melegari, M.; Rindhe, S.; Mitsche, M.; Yang, C.; Batten, K.; Huffman, K.E.; Liu, J.; et al. Fatty acid oxidation mediated by acyl-CoA synthetase long chain 3 is required for mutant kras lung tumorigenesis. Cell Rep. 2016, 16, 1614–1628. [Google Scholar]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mtorc1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [PubMed]
- Schaffer, J.E.; Lodish, H.F. Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 1994, 79, 427–436. [Google Scholar] [PubMed]
- Schwenk, R.W.; Holloway, G.P.; Luiken, J.J.; Bonen, A.; Glatz, J.F. Fatty acid transport across the cell membrane: Regulation by fatty acid transporters. Prostaglandins Leukot Essent Fatty Acids 2010, 82, 149–154. [Google Scholar] [PubMed]
- Sanchez-Martinez, R.; Cruz-Gil, S.; Garcia-Alvarez, M.S.; Reglero, G.; Ramirez de Molina, A. Complementary acsl isoforms contribute to a non-warburg advantageous energetic status characterizing invasive colon cancer cells. Sci. Rep. 2017, 7, 11143. [Google Scholar] [PubMed]
- Park, S.; Oh, J.; Kim, M.; Jin, E.J. Bromelain effectively suppresses kras-mutant colorectal cancer by stimulating ferroptosis. Anim. Cells Syst. (Seoul) 2018, 22, 334–340. [Google Scholar] [PubMed]
- Wright, H.J.; Hou, J.; Xu, B.; Cortez, M.; Potma, E.O.; Tromberg, B.J.; Razorenova, O.V. Cdcp1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl. Acad. Sci. USA 2017, 114, E6556–E6565. [Google Scholar] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [PubMed]
- Chen, W.C.; Wang, C.Y.; Hung, Y.H.; Weng, T.Y.; Yen, M.C.; Lai, M.D. Systematic analysis of gene expression alterations and clinical outcomes for long-chain acyl-coenzyme a synthetase family in cancer. PLoS ONE 2016, 11, e0155660. [Google Scholar]
- Li, L.O.; Klett, E.L.; Coleman, R.A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 246–251. [Google Scholar]
- Bauer, P.V.; Duca, F.A.; Waise, T.M.Z.; Dranse, H.J.; Rasmussen, B.A.; Puri, A.; Rasti, M.; O’Brien, C.A.; Lam, T.K.T. Lactobacillus gasseri in the upper small intestine impacts an acsl3-dependent fatty acid-sensing pathway regulating whole-body glucose homeostasis. Cell Metab. 2018, 27, 572–587. [Google Scholar]
- Ellis, J.M.; Frahm, J.L.; Li, L.O.; Coleman, R.A. Acyl-coenzyme a synthetases in metabolic control. Curr. Opin. Lipidol. 2010, 21, 212–217. [Google Scholar] [PubMed]
- Kang, M.J.; Fujino, T.; Sasano, H.; Minekura, H.; Yabuki, N.; Nagura, H.; Iijima, H.; Yamamoto, T.T. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. USA 1997, 94, 2880–2884. [Google Scholar] [PubMed]
- Fujino, T.; Kang, M.J.; Suzuki, H.; Iijima, H.; Yamamoto, T. Molecular characterization and expression of rat acyl-CoA synthetase 3. J. Biol. Chem. 1996, 271, 16748–16752. [Google Scholar] [PubMed]
- Oikawa, E.; Iijima, H.; Suzuki, T.; Sasano, H.; Sato, H.; Kamataki, A.; Nagura, H.; Kang, M.J.; Fujino, T.; Suzuki, H.; et al. A novel acyl-CoA synthetase, acs5, expressed in intestinal epithelial cells and proliferating preadipocytes. J. Biochem. 1998, 124, 679–685. [Google Scholar] [PubMed]
- Mashek, D.G.; Bornfeldt, K.E.; Coleman, R.A.; Berger, J.; Bernlohr, D.A.; Black, P.; DiRusso, C.C.; Farber, S.A.; Guo, W.; Hashimoto, N.; et al. Revised nomenclature for the mammalian long-chain acyl-CoA synthetase gene family. J. Lipid Res. 2004, 45, 1958–1961. [Google Scholar] [PubMed]
- Kanter, J.E.; Tang, C.; Oram, J.F.; Bornfeldt, K.E. Acyl-CoA synthetase 1 is required for oleate and linoleate mediated inhibition of cholesterol efflux through atp-binding cassette transporter a1 in macrophages. Biochim. Biophys. Acta. 2012, 1821, 358–364. [Google Scholar]
- Klett, E.L.; Chen, S.; Yechoor, A.; Lih, F.B.; Coleman, R.A. Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J. Lipid Res. 2017, 58, 884–894. [Google Scholar]
- Lopes-Marques, M.; Cunha, I.; Reis-Henriques, M.A.; Santos, M.M.; Castro, L.F. Diversity and history of the long-chain acyl-CoA synthetase (acsl) gene family in vertebrates. BMC Evol. Biol. 2013, 13, 271. [Google Scholar]
- Spector, A.A.; Yorek, M.A. Membrane lipid composition and cellular function. J. Lipid Res. 1985, 26, 1015–1035. [Google Scholar]
- Yamashita, A.; Hayashi, Y.; Nemoto-Sasaki, Y.; Ito, M.; Oka, S.; Tanikawa, T.; Waku, K.; Sugiura, T. Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms. Prog. Lipid Res. 2014, 53, 18–81. [Google Scholar]
- Kassan, A.; Herms, A.; Fernandez-Vidal, A.; Bosch, M.; Schieber, N.L.; Reddy, B.J.; Fajardo, A.; Gelabert-Baldrich, M.; Tebar, F.; Enrich, C.; et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in er microdomains. J. Cell Biol. 2013, 203, 985–1001. [Google Scholar] [PubMed]
- Fujimoto, Y.; Itabe, H.; Kinoshita, T.; Homma, K.J.; Onoduka, J.; Mori, M.; Yamaguchi, S.; Makita, M.; Higashi, Y.; Yamashita, A.; et al. Involvement of acsl in local synthesis of neutral lipids in cytoplasmic lipid droplets in human hepatocyte huh7. J. Lipid Res. 2007, 48, 1280–1292. [Google Scholar] [PubMed]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by pten loss and PI3k/Akt activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [PubMed]
- Petan, T.; Jarc, E.; Jusovic, M. Lipid droplets in cancer: Guardians of fat in a stressful world. Molecules 2018, 23, 1941. [Google Scholar]
- Ackerman, D.; Tumanov, S.; Qiu, B.; Michalopoulou, E.; Spata, M.; Azzam, A.; Xie, H.; Simon, M.C.; Kamphorst, J.J. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. 2018, 24, 2596–2605.e5. [Google Scholar] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by hif-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [PubMed]
- Durgan, D.J.; Smith, J.K.; Hotze, M.A.; Egbejimi, O.; Cuthbert, K.D.; Zaha, V.G.; Dyck, J.R.; Abel, E.D.; Young, M.E. Distinct transcriptional regulation of long-chain acyl-CoA synthetase isoforms and cytosolic thioesterase 1 in the rodent heart by fatty acids and insulin. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2480–H2497. [Google Scholar] [PubMed]
- Poppelreuther, M.; Rudolph, B.; Du, C.; Grossmann, R.; Becker, M.; Thiele, C.; Ehehalt, R.; Fullekrug, J. The n-terminal region of acyl-CoA synthetase 3 is essential for both the localization on lipid droplets and the function in fatty acid uptake. J. Lipid Res. 2012, 53, 888–900. [Google Scholar]
- Ansari, I.H.; Longacre, M.J.; Stoker, S.W.; Kendrick, M.A.; O’Neill, L.M.; Zitur, L.J.; Fernandez, L.A.; Ntambi, J.M.; MacDonald, M.J. Characterization of acyl-CoA synthetase isoforms in pancreatic beta cells: Gene silencing shows participation of acsl3 and acsl4 in insulin secretion. Arch. Biochem. Biophys. 2017, 618, 32–43. [Google Scholar]
- Obata, Y.; Fukumoto, Y.; Nakayama, Y.; Kuga, T.; Dohmae, N.; Yamaguchi, N. The lyn kinase c-lobe mediates golgi export of lyn through conformation-dependent acsl3 association. J. Cell Sci. 2010, 123, 2649–2662. [Google Scholar]
- Liu, Z.; Huang, Y.; Hu, W.; Huang, S.; Wang, Q.; Han, J.; Zhang, Y.Q. Dacsl, the drosophila ortholog of acyl-CoA synthetase long-chain family member 3 and 4, inhibits synapse growth by attenuating bone morphogenetic protein signaling via endocytic recycling. J. Neurosci. 2014, 34, 2785–2796. [Google Scholar]
- Lewin, T.M.; Kim, J.H.; Granger, D.A.; Vance, J.E.; Coleman, R.A. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 2001, 276, 24674–24679. [Google Scholar]
- Kuch, E.M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brugger, B.; Sreemmel, W.; Ehehalt, R.; Poppelreuther, M.; Fullekrug, J. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 2014, 1841, 227–239. [Google Scholar]
- Meller, N.; Morgan, M.E.; Wong, W.P.; Altemus, J.B.; Sehayek, E. Targeting of acyl-CoA synthetase 5 decreases jejunal fatty acid activation with no effect on dietary long-chain fatty acid absorption. Lipids Health Dis. 2013, 12, 88. [Google Scholar]
- Fujino, T.; Yamamoto, T. Cloning and functional expression of a novel long-chain acyl-CoA synthetase expressed in brain. J. Biochem. 1992, 111, 197–203. [Google Scholar]
- Sanchez-Martinez, R.; Cruz-Gil, S.; Gomez de Cedron, M.; Alvarez-Fernandez, M.; Vargas, T.; Molina, S.; Garcia, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar]
- Cruz-Gil, S.; Sanchez-Martinez, R.; Gomez de Cedron, M.; Martin-Hernandez, R.; Vargas, T.; Molina, S.; Herranz, J.; Davalos, A.; Reglero, G.; Ramirez de Molina, A. Targeting the lipid metabolic axis acsl/scd in colorectal cancer progression by therapeutic mirnas: Mir-19b-1 role. J. Lipid Res. 2018, 59, 14–24. [Google Scholar]
- Gassler, N.; Herr, I.; Schneider, A.; Penzel, R.; Langbein, L.; Schirmacher, P.; Kopitz, J. Impaired expression of acyl-CoA synthetase 5 in sporadic colorectal adenocarcinomas. J. Pathol. 2005, 207, 295–300. [Google Scholar]
- Hartmann, F.; Sparla, D.; Tute, E.; Tamm, M.; Schneider, U.; Jeon, M.K.; Kasperk, R.; Gassler, N.; Kaemmerer, E. Low acyl-CoA synthetase 5 expression in colorectal carcinomas is prognostic for early tumour recurrence. Pathol. Res. Pract. 2017, 213, 261–266. [Google Scholar]
- Wilson, K.E.; Bachawal, S.V.; Tian, L.; Willmann, J.K. Multiparametric spectroscopic photoacoustic imaging of breast cancer development in a transgenic mouse model. Theranostics 2014, 4, 1062–1071. [Google Scholar]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for myc-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar]
- Yen, M.C.; Kan, J.Y.; Hsieh, C.J.; Kuo, P.L.; Hou, M.F.; Hsu, Y.L. Association of long-chain acyl-coenzyme a synthetase 5 expression in human breast cancer by estrogen receptor status and its clinical significance. Oncol. Rep. 2017, 37, 3253–3260. [Google Scholar]
- Wang, Y.; Cai, X.; Zhang, S.; Cui, M.; Liu, F.; Sun, B.; Zhang, W.; Zhang, X.; Ye, L. Hbxip up-regulates acsl1 through activating transcriptional factor sp1 in breast cancer. Biochem. Biophys. Res. Commun. 2017, 484, 565–571. [Google Scholar]
- Wang, J.; Scholtens, D.; Holko, M.; Ivancic, D.; Lee, O.; Hu, H.; Chatterton, R.T., Jr.; Sullivan, M.E.; Hansen, N.; Bethke, K.; et al. Lipid metabolism genes in contralateral unaffected breast and estrogen receptor status of breast cancer. Cancer Prev. Res. (Phila) 2013, 6, 321–330. [Google Scholar]
- Wu, X.; Li, Y.; Wang, J.; Wen, X.; Marcus, M.T.; Daniels, G.; Zhang, D.Y.; Ye, F.; Wang, L.H.; Du, X.; et al. Long chain fatty acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS ONE 2013, 8, e77060. [Google Scholar]
- Orlando, U.D.; Castillo, A.F.; Medrano, M.A.R.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4 is implicated in drug resistance in breast cancer cell lines involving the regulation of energy-dependent transporter expression. Biochem. Pharmacol. 2019, 159, 52–63. [Google Scholar]
- Migita, T.; Takayama, K.I.; Urano, T.; Obinata, D.; Ikeda, K.; Soga, T.; Takahashi, S.; Inoue, S. Acsl3 promotes intratumoral steroidogenesis in prostate cancer cells. Cancer Sci. 2017, 108, 2011–2021. [Google Scholar]
- Saab, J.; Santos-Zabala, M.L.; Loda, M.; Stack, E.C.; Hollmann, T.J. Fatty acid synthase and acetyl-CoA carboxylase are expressed in nodal metastatic melanoma but not in benign intracapsular nodal nevi. Am. J. Dermatopathol. 2018, 40, 259–264. [Google Scholar]
- Giampietri, C.; Petrungaro, S.; Cordella, M.; Tabolacci, C.; Tomaipitinca, L.; Facchiano, A.; Eramo, A.; Filippini, A.; Facchiano, F.; Ziparo, E. Lipid storage and autophagy in melanoma cancer cells. Int. J. Mol. Sci. 2017, 18, 1271. [Google Scholar]
- Shyu, P., Jr.; Wong, X.F.A.; Crasta, K.; Thibault, G. Dropping in on lipid droplets: Insights into cellular stress and cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Yao, H.; Ye, J. Long chain acyl-CoA synthetase 3-mediated phosphatidylcholine synthesis is required for assembly of very low density lipoproteins in human hepatoma huh7 cells. J. Biol. Chem. 2008, 283, 849–854. [Google Scholar]
- Young, P.A.; Senkal, C.E.; Suchanek, A.L.; Grevengoed, T.J.; Lin, D.D.; Zhao, L.; Crunk, A.E.; Klett, E.L.; Fullekrug, J.; Obeid, L.M.; et al. Long-chain acyl-CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J. Biol. Chem. 2018, 293, 16724–16740. [Google Scholar]
- Nwosu, Z.C.; Megger, D.A.; Hammad, S.; Sitek, B.; Roessler, S.; Ebert, M.P.; Meyer, C.; Dooley, S. Identification of the consistently altered metabolic targets in human hepatocellular carcinoma. Cell Mol. Gastroenterol. Hepatol. 2017, 4, 303–323. [Google Scholar]
- Liang, Y.C.; Wu, C.H.; Chu, J.S.; Wang, C.K.; Hung, L.F.; Wang, Y.J.; Ho, Y.S.; Chang, J.G.; Lin, S.Y. Involvement of fatty acid-CoA ligase 4 in hepatocellular carcinoma growth: Roles of cyclic amp and p38 mitogen-activated protein kinase. World J. Gastroenterol. 2005, 11, 2557–2563. [Google Scholar]
- Xu, C.; Wang, G.; Hao, Y.; Zhi, J.; Zhang, L.; Chang, C. Correlation analysis between gene expression profile of rat liver tissues and high-fat emulsion-induced nonalcoholic fatty liver. Dig. Dis. Sci. 2011, 56, 2299–2308. [Google Scholar]
- Issa, D.; Alkhouri, N. Nonalcoholic fatty liver disease and hepatocellular carcinoma: New insights on presentation and natural history. Hepatobiliary Surg. Nutr. 2017, 6, 401–403. [Google Scholar]
- Radif, Y.; Ndiaye, H.; Kalantzi, V.; Jacobs, R.; Hall, A.; Minogue, S.; Waugh, M.G. The endogenous subcellular localisations of the long chain fatty acid-activating enzymes acsl3 and acsl4 in sarcoma and breast cancer cells. Mol. Cell. Biochem. 2018, 448, 275–286. [Google Scholar]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable isotope labeling highlights enhanced fatty acid and lipid metabolism in human acute myeloid leukemia. Int. J. Mol. Sci. 2018, 19, 3325. [Google Scholar]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone marrow adipocytes facilitate fatty acid oxidation activating ampk and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar]
- Tomoda, H.; Igarashi, K.; Cyong, J.C.; Omura, S. Evidence for an essential role of long chain acyl-CoA synthetase in animal cell proliferation. Inhibition of long chain acyl-CoA synthetase by triacsins caused inhibition of raji cell proliferation. J. Biol Chem 1991, 266, 4214–4219. [Google Scholar]
- Kaemmerer, E.; Peuscher, A.; Reinartz, A.; Liedtke, C.; Weiskirchen, R.; Kopitz, J.; Gassler, N. Human intestinal acyl-CoA synthetase 5 is sensitive to the inhibitor triacsin c. World J. Gastroenterol 2011, 17, 4883–4889. [Google Scholar] [PubMed]
- Kim, Y.; George, D.; Prior, A.M.; Prasain, K.; Hao, S.; Le, D.D.; Hua, D.H.; Chang, K.O. Novel triacsin c analogs as potential antivirals against rotavirus infections. Eur. J. Med. Chem. 2012, 50, 311–318. [Google Scholar] [PubMed]
- Prior, A.M.; Zhang, M.; Blakeman, N.; Datta, P.; Pham, H.; Chen, Q.; Young, L.H.; Weis, M.T.; Hua, D.H. Inhibition of long chain fatty acyl-CoA synthetase (acsl) and ischemia reperfusion injury. Bioorg. Med. Chem. Lett. 2014, 24, 1057–1061. [Google Scholar] [PubMed]
- Recuero-Checa, M.A.; Sharma, M.; Lau, C.; Watkins, P.A.; Gaydos, C.A.; Dean, D. Chlamydia trachomatis growth and development requires the activity of host long-chain acyl-CoA synthetases (acsls). Sci. Rep. 2016, 6, 23148. [Google Scholar] [PubMed]
- Cao, A.; Li, H.; Zhou, Y.; Wu, M.; Liu, J. Long chain acyl-CoA synthetase-3 is a molecular target for peroxisome proliferator-activated receptor delta in hepg2 hepatoma cells. J. Biol. Chem. 2010, 285, 16664–16674. [Google Scholar]
- Burton, J.D.; Goldenberg, D.M.; Blumenthal, R.D. Potential of peroxisome proliferator-activated receptor gamma antagonist compounds as therapeutic agents for a wide range of cancer types. PPAR Res. 2008, 2008, 494161. [Google Scholar] [PubMed]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of acsl4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar]
- Mashima, T.; Sato, S.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Promotion of glioma cell survival by acyl-CoA synthetase 5 under extracellular acidosis conditions. Oncogene 2009, 28, 9–19. [Google Scholar]
- Mashima, T.; Sato, S.; Okabe, S.; Miyata, S.; Matsuura, M.; Sugimoto, Y.; Tsuruo, T.; Seimiya, H. Acyl-CoA synthetase as a cancer survival factor: Its inhibition enhances the efficacy of etoposide. Cancer Sci. 2009, 100, 1556–1562. [Google Scholar]
- Cotte, A.K.; Aires, V.; Fredon, M.; Limagne, E.; Derangere, V.; Thibaudin, M.; Humblin, E.; Scagliarini, A.; de Barros, J.P.; Hillon, P.; et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 2018, 9, 322. [Google Scholar]
- Liu, K.T.; Yeh, I.J.; Chou, S.K.; Yen, M.C.; Kuo, P.L. Regulatory mechanism of fatty acidcoa metabolic enzymes under endoplasmic reticulum stress in lung cancer. Oncol. Rep. 2018, 40, 2674–2682. [Google Scholar] [PubMed]
- Guha, P.; Kaptan, E.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mtorc1. Oncotarget 2017, 8, 68191–68207. [Google Scholar] [PubMed]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates er stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting n-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [PubMed]
- Wang, X.; Xiong, W.; Tang, Y. Tunicamycin suppresses breast cancer cell growth and metastasis via regulation of the protein kinase b/nuclear factor-kappab signaling pathway. Oncol. Lett. 2018, 15, 4137–4142. [Google Scholar] [PubMed]
- Ramadori, G.; Konstantinidou, G.; Venkateswaran, N.; Biscotti, T.; Morlock, L.; Galie, M.; Williams, N.S.; Luchetti, M.; Santinelli, A.; Scaglioni, P.P.; et al. Diet-induced unresolved er stress hinders kras-driven lung tumorigenesis. Cell Metab. 2015, 21, 117–125. [Google Scholar] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. Acsl4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell. Biol. 2016, 26, 165–176. [Google Scholar]
- Taylor, W.R.; Fedorka, S.R.; Gad, I.; Shah, R.; Alqahtani, H.D.; Koranne, R.; Kuganesan, N.; Dlamini, S.; Rogers, T.; Al-Hamashi, A.; et al. Small-molecule ferroptotic agents with potential to selectively target cancer stem cells. Sci. Rep. 2019, 9, 5926. [Google Scholar]
- Bowman, T.A.; O’Keeffe, K.R.; D’Aquila, T.; Yan, Q.W.; Griffin, J.D.; Killion, E.A.; Salter, D.M.; Mashek, D.G.; Buhman, K.K.; Greenberg, A.S. Acyl coa synthetase 5 (acsl5) ablation in mice increases energy expenditure and insulin sensitivity and delays fat absorption. Mol. Metab. 2016, 5, 210–220. [Google Scholar]
- Fernandez, R.F.; Kim, S.Q.; Zhao, Y.; Foguth, R.M.; Weera, M.M.; Counihan, J.L.; Nomura, D.K.; Chester, J.A.; Cannon, J.R.; Ellis, J.M. Acyl-CoA synthetase 6 enriches the neuroprotective omega-3 fatty acid dha in the brain. Proc. Natl. Acad. Sci. USA 2018, 115, 12525–12530. [Google Scholar] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [PubMed]


{kind=link}
| Target Isoenzyme | Cancer Type | Effect | Reference |
|---|---|---|---|
| ACSL1 | CRC cell line | Overexpression: enhanced wound healing, invasion, proliferation, glycolysis and EMT (this latter, only when combined with ACSL4 and stearoyl-CoA desaturase (SCD1) overexpression). Knockdown: decreased cell proliferation, migration, and anchorage-independent growth. miR-19b-1 expression: decreased invasion and proliferation. | [24,28,56,57] |
| BC cell lines | Knockdown: decreased proliferation, colony formation and cell viability. | [28] | |
| NSCLC cell lines | Knockdown: enhanced proliferation and invasiveness of non-small cell lung cancer cell lines. | [28] | |
| ACSL3 | BC cell lines | Knockdown: decreased proliferation and viability, increased FAO/decreased lipid droplets content. | [26] |
| PC cell lines | Overexpression: protection against ER stress inducers. Increased intracellular steroidogenesis. | [67] | |
| NSCLC cell lines | Knockdown: decreased cell proliferation, colony formation and reduced FAO. | [20] | |
| HCC | Knockdown: decreased very low density lipoprotein (VLDL) secretion. | [72] | |
| KRAS NSCLC GEM mouse model | Total mouse knockout: reduced tumor initiation/tumor burden. | [20] | |
| ACSL4 | CRC cell line | Overexpression: enhanced wound healing, invasion, proliferation and glycolysis. Knockdown: decreased cell proliferation and glycolysis, insensitivity to ferroptosis inducers. miR-19b-1 expression: decreased invasion and proliferation. | [24,56,57,87] |
| HCC cell lines | Knockdown: reduced proliferation. | [74] | |
| ER+ BC cell lines | Overexpression: enhanced cell proliferation, increased drug efflux and chemotherapy resistance. | [65,66] | |
| Quadruple-negative BC cell lines | Overexpression: Increased invasion and anchorage independent growth in vitro and increased tumor burden in vivo. | [65,66] | |
| ACSL5 | Glioma cell lines | Overexpression: enhanced cell survival under extracellular acidosis. Knockdown: reduced cell survival under extracellular acidosis. | [88] |
| ACSL6 | NA | NA | NA |
| ACSL1, 3, 4, (5) | ER+, ER− and BC cell lines | Triacsin C: decreased proliferation and decreased chemotherapy resistance. | [62,65] |
| CRC cell lines | Triacsin C: enhanced sensitization to stearoyl-CoA desaturase (SCD1) inhibition treatment. | [56] | |
| Glioma xenograft mouse model | Triacsin C: sensitization to low dose etoposide treatment. | [89] | |
| CRC xenograft mouse model | Triacsin C: decreased lipid droplets content and increased sensitivity to chemotherapy. | [90] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi Sebastiano, M.; Konstantinidou, G. Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 3624. https://doi.org/10.3390/ijms20153624
Rossi Sebastiano M, Konstantinidou G. Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. International Journal of Molecular Sciences. 2019; 20(15):3624. https://doi.org/10.3390/ijms20153624
Chicago/Turabian StyleRossi Sebastiano, Matteo, and Georgia Konstantinidou. 2019. "Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy" International Journal of Molecular Sciences 20, no. 15: 3624. https://doi.org/10.3390/ijms20153624
APA StyleRossi Sebastiano, M., & Konstantinidou, G. (2019). Targeting Long Chain Acyl-CoA Synthetases for Cancer Therapy. International Journal of Molecular Sciences, 20(15), 3624. https://doi.org/10.3390/ijms20153624