Transactivation Function-1-Mediated Partial Agonist Activity of Selective Estrogen Receptor Modulator Requires Homo-Dimerization of the Estrogen Receptor α Ligand Binding Domain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
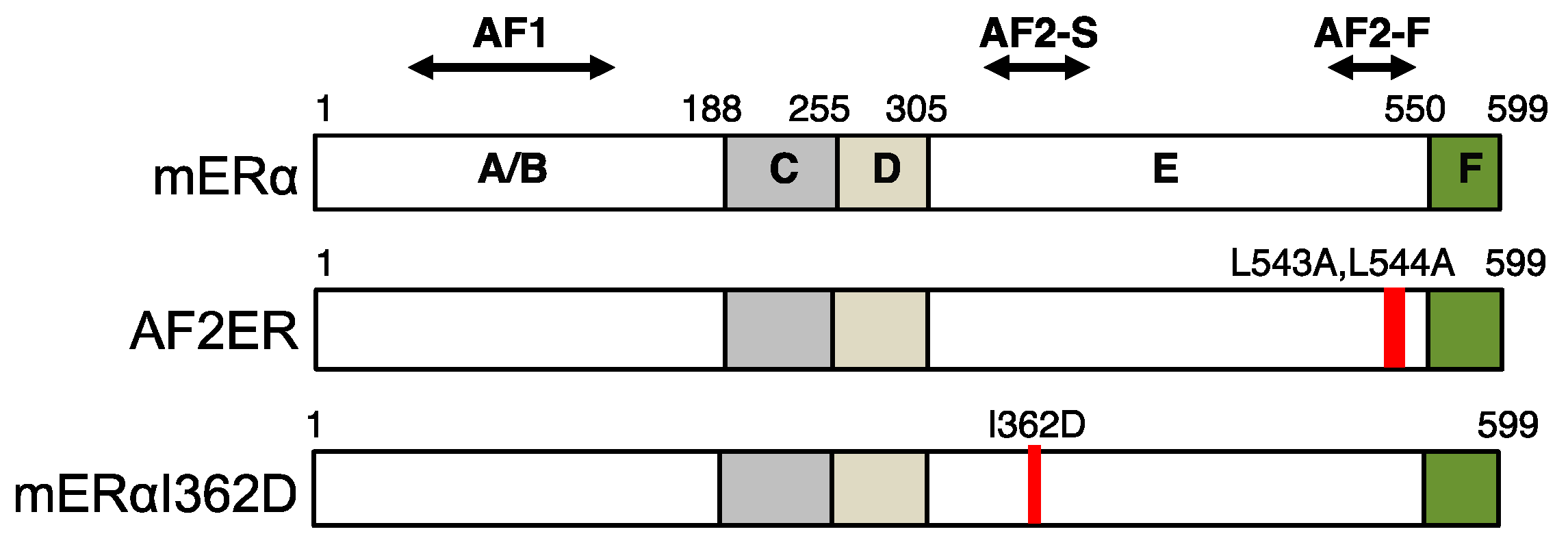
2. The Mutation of Helix 12 Reverses Antagonistic Chemicals to Agonists
3. The Ligand-Dependent Regulation of ERα AF-1 Activity
4. The F-Domain Functionality for the SERM-Dependent Partial Agonist Activity
5. Conclusions
6. Summary
- Helix 12 of ERα LBD defines ligands as agonists or antagonists. Antagonists can work as agonists through ERα mutants with a disrupted helix 12 and that transcriptional activity is derived from AF-1.
- Helix 3 of ERα LBD contributes to the attenuation of AF-1 activity. Anomalous charge on helix 3 disrupts the LBD functions of AF-1 suppression and AF-2 transactivation.
- SERMs induce ERα homo-dimerization through the LBD without recruitment of AF-2 coactivators. The AF-1-dependent transcriptional activity of SERMs correlates with the activity of LBD homo-dimerization.
- The F-domain of ERα contributes to the SERM (4OHT)-dependent LBD homo-dimerization.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERα | Estrogen receptor alpha |
| SERM | Selective estrogen receptor modulator |
| DBD | DNA binding domain |
| LBD | Ligand binding domain |
| KO | Knockout |
| 4OHT | 4-hydroxytamoxifen |
| SERD | Selective estrogen receptor degraders |
| E2 | 17β-estradiol |
| Vit | Vitellogenin |
| M2H | Mammalian two-hybrid |
| DES | Diethylstilbestrol |
| ERE | Estrogen responsive element |
| Gal4RE | Gal4 responsive element |
References
- Greene, G.L.; Gilna, P.; Waterfield, M.; Baker, A.; Hort, Y.; Shine, J. Sequence and expression of human estrogen receptor complementary DNA. Science 1986, 231, 1150–1154. [Google Scholar] [CrossRef] [PubMed]
- White, R.; Lees, J.A.; Needham, M.; Ham, J.; Parker, M. Structural Organization and Expression of the Mouse Estrogen Receptor. Mol. Endocrinol. 1987, 1, 735–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, S.; Sakai, M.; Muramatsu, M. Molecular cloning and characterization of rat estrogen receptor cDNA. Nucleic Acids Res. 1987, 15, 2499–2513. [Google Scholar] [CrossRef] [PubMed]
- Krust, A.; Green, S.; Argos, P.; Kumar, V.; Walter, P.; Bornert, J.M.; Chambon, P. The chicken oestrogen receptor sequence: Homology with v-erbA and the human oestrogen and glucocorticoid receptors. EMBO J. 1986, 5, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chandra, V.; Rastinejad, F. Structural Overview of the Nuclear Receptor Superfamily: Insights into Physiology and Therapeutics. Annu. Rev. Physiol. 2010, 72, 247–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, R.M.; Mangelsdorf, D.J. Nuclear Receptors, RXR, and the Big Bang. Cell 2014, 157, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronemeyer, H.; Gustafsson, J.-Å.; Laudet, V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004, 3, 950–964. [Google Scholar] [CrossRef]
- Arao, Y.; Hamilton, K.J.; Ray, M.K.; Scott, G.; Mishina, Y.; Korach, K.S. Estrogen receptor α AF-2 mutation results in antagonist reversal and reveals tissue selective function of estrogen receptor modulators. Proc. Natl. Acad. Sci. USA 2011, 108, 14986–14991. [Google Scholar] [CrossRef]
- Arao, Y.; Hamilton, K.J.; Korach, K.S. The Transactivating Function 2 (AF-2) of Estrogen Receptor (ER) α Is Indispensable for ERα-Mediated Physiological Responses and AF-1 Activity. OJEMD 2013, 3, 12–19. [Google Scholar] [CrossRef]
- Montano, M.M.; Ekena, K.; Krueger, K.D.; Keller, A.L.; Katzenellenbogen, B.S. Human estrogen receptor ligand activity inversion mutants: Receptors that interpret antiestrogens as estrogens and estrogens as antiestrogens and discriminate among different antiestrogens. Mol. Endocrinol. 1996, 10, 230–242. [Google Scholar]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Bunone, G.; Briand, P.A.; Miksicek, R.J.; Picard, D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phophorylation. EMBO J. 1996, 15, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Rogatsky, I.; Trowbridge, J.M.; Garabedian, M.J. Potentiation of human estrogen receptor alpha transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J. Biol. Chem. 1999, 274, 22296–22302. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Riedl, T.; Washbrook, E.; Pace, P.E.; Coombes, R.C.; Egly, J.M.; Ali, S. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 2000, 6, 127–137. [Google Scholar] [CrossRef]
- Curtis, S.W.; Washburn, T.; Sewall, C.; DiAugustine, R.; Lindzey, J.; Couse, J.F.; Korach, K.S. Physiological coupling of growth factor and steroid receptor signaling pathways: Estrogen receptor knockout mice lack estrogen-like response to epidermal growth factor. Proc. Natl. Acad. Sci. USA 1996, 93, 12626–12630. [Google Scholar] [CrossRef]
- Arao, Y.; Coons, L.A.; Zuercher, W.J.; Korach, K.S. Transactivation Function-2 of Estrogen Receptor α Contains Transactivation Function-1-regulating Element. J. Biol. Chem. 2015, 290, 17611–17627. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.R.; Skafar, D.F. Modulation of nuclear receptor activity by the F domain. Mol. Cell. Endocrinol. 2015, 418, 298–305. [Google Scholar] [CrossRef]
- Kaminetsky, J.; Hemani, M.L. Clomiphene citrate and enclomiphene for the treatment of hypogonadal androgen deficiency. Expert Opin. Investig. Drugs 2009, 18, 1947–1955. [Google Scholar] [CrossRef]
- Ahmad, A.; Shahabuddin, S.; Sheikh, S.; Kale, P.; Krishnappa, M.; Rane, R.C.; Ahmad, I. Endoxifen, a New Cornerstone of Breast Cancer Therapy: Demonstration of Safety, Tolerability, and Systemic Bioavailability in Healthy Human Subjects. Clin. Pharmacol. Ther. 2009, 88, 814–817. [Google Scholar] [CrossRef]
- Ali, S.; Buluwela, L.; Coombes, R.C. Antiestrogens and Their Therapeutic Applications in Breast Cancer and Other Diseases. Annu. Rev. Med. 2011, 62, 217–232. [Google Scholar] [CrossRef] [Green Version]
- Chi, F.; Wu, R.; Zeng, Y.; Xing, R.; Liu, Y.; Xu, Z. Effects of toremifene versus tamoxifen on breast cancer patients: A meta-analysis. Breast Cancer 2012, 20, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, J.V.; Pickar, J.H.; Racketa, J.; Mirkin, S. Bazedoxifene/conjugated estrogens for menopausal symptom treatment and osteoporosis prevention. Climacteric 2012, 15, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Elkinson, S.; Yang, L.P.H. Ospemifene: First Global Approval. Drugs 2013, 73, 605–612. [Google Scholar] [CrossRef] [PubMed]
- D’Amelio, P.; Isaia, G.C. The use of raloxifene in osteoporosis treatment. Expert Opin. Pharmacother. 2013, 14, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Johnston, M.A.; Capers, C.; Braccia, D. Toremifene for Breast Cancer: A Review of 20 Years of Data. Clin. Breast Cancer 2014, 14, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: Three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 2000, 21, 381–388. [Google Scholar] [CrossRef]
- Mak, H.Y.; Hoare, S.; Henttu, P.M.; Parker, M.G. Molecular determinants of the estrogen receptor-coactivator interface. Mol. Cell. Biol. 1999, 19, 3895–3903. [Google Scholar] [CrossRef]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927–937. [Google Scholar] [CrossRef]
- Pike, A.C.; Brzozowski, A.M.; Walton, J.; Hubbard, R.E.; Thorsell, A.G.; Li, Y.L.; Gustafsson, J.A.; Carlquist, M. Structural insights into the mode of action of a pure antiestrogen. Structure 2001, 9, 145–153. [Google Scholar] [CrossRef]
- Lonard, D.M.; Nawaz, Z.; Smith, C.L.; O’Malley, B.W. The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol. Cell 2000, 5, 939–948. [Google Scholar] [CrossRef]
- Dauvois, S.; Danielian, P.S.; White, R.; Parker, M.G. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl. Acad. Sci. USA 1992, 89, 4037–4041. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Hamilton, K.J.; Goulding, E.H.; Janardhan, K.S.; Eddy, E.M.; Korach, K.S. Transactivating function (AF) 2-mediated AF-1 activity of estrogen receptor α is crucial to maintain male reproductive tract function. Proc. Natl. Acad. Sci. USA 2012, 109, 21140–21145. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Hamilton, K.J.; Lierz, S.L.; Korach, K.S. N-terminal transactivation function, AF-1, of estrogen receptor alpha controls obesity through enhancement of energy expenditure. Mol. Metab. 2018, 18, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Hamilton, K.J.; Coons, L.A.; Korach, K.S. Estrogen receptor α L543A, L544A mutation changes antagonists to agonists, correlating with the ligand binding domain dimerization associated with DNA binding activity. J. Biol. Chem. 2013, 288, 21105–21116. [Google Scholar] [CrossRef] [PubMed]
- Mahfoudi, A.; Roulet, E.; Dauvois, S.; Parker, M.G.; Wahli, W. Specific mutations in the estrogen receptor change the properties of antiestrogens to full agonists. Proc. Natl. Acad. Sci. USA 1995, 92, 4206–4210. [Google Scholar] [CrossRef] [PubMed]
- Fawell, S.E.; Lees, J.A.; White, R.; Parker, M.G. Characterization and colocalization of steroid binding and dimerization activities in the mouse estrogen receptor. Cell 1990, 60, 953–962. [Google Scholar] [CrossRef]
- Billon-Galés, A.; Krust, A.; Fontaine, C.; Abot, A.; Flouriot, G.; Toutain, C.; Bergès, H.; Gadeau, A.-P.; Lenfant, F.; Gourdy, P.; et al. Activation function 2 (AF2) of estrogen receptor-alpha is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc. Natl. Acad. Sci. USA 2011, 108, 13311–13316. [Google Scholar] [CrossRef] [PubMed]
- Handgraaf, S.; Riant, E.; Fabre, A.; Waget, A.; Burcelin, R.; Lière, P.; Krust, A.; Chambon, P.; Arnal, J.-F.; Gourdy, P. Prevention of obesity and insulin resistance by estrogens requires ERα activation function-2 (ERαAF-2), whereas ERαAF-1 is dispensable. Diabetes 2013, 62, 4098–4108. [Google Scholar] [CrossRef] [PubMed]
- Movérare-Skrtic, S.; Börjesson, A.E.; Farman, H.H.; Sjögren, K.; Windahl, S.H.; Lagerquist, M.K.; Andersson, A.; Stubelius, A.; Carlsten, H.; Gustafsson, J.-Å.; et al. The estrogen receptor antagonist ICI 182,780 can act both as an agonist and an inverse agonist when estrogen receptor α AF-2 is modified. Proc. Natl. Acad. Sci. USA 2014, 111, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Métivier, R.; Stark, A.; Flouriot, G.; Hübner, M.R.; Brand, H.; Penot, G.; Manu, D.; Denger, S.; Reid, G.; Kos, M.; et al. A dynamic structural model for estrogen receptor-alpha activation by ligands, emphasizing the role of interactions between distant A and E domains. Mol. Cell 2002, 10, 1019–1032. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kitamoto, T.; Masuhiro, Y.; Watanabe, M.; Kase, T.; Metzger, D.; Yanagisawa, J.; Kato, S. p300 mediates functional synergism between AF-1 and AF-2 of estrogen receptor alpha and beta by interacting directly with the N-terminal A/B domains. J. Biol. Chem. 2000, 275, 15645–15651. [Google Scholar] [CrossRef] [PubMed]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.E.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Metzger, D.; Chambon, P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J. 1990, 9, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, D. The Molecular Pharmacology of SERMs. Trends Endocrinol. Metab. 1999, 10, 301–311. [Google Scholar] [CrossRef]
- Fontaine, C.; Abot, A.; Billon-Galés, A.; Flouriot, G.; Bergès, H.; Grunenwald, E.; Vinel, A.; Valera, M.-C.; Gourdy, P.; Arnal, J.-F. Tamoxifen Elicits Atheroprotection through Estrogen Receptor alpha AF-1 But Does Not Accelerate Reendothelialization. Am. J. Pathol. 2013, 183, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Arao, Y.; Korach, K.S. Detecting the Ligand-binding Domain Dimerization Activity of Estrogen Receptor Alpha Using the Mammalian Two-Hybrid Assay. J. Vis. Exp. 2018, e58758. [Google Scholar] [CrossRef] [PubMed]
- Eiler, S.; Gangloff, M.; Duclaud, S.; Moras, D.; Ruff, M. Overexpression, purification, and crystal structure of native ER alpha LBD. Protein Expr. Purif. 2001, 22, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.H.; Markaverich, B.M. The agonistic-antagonistic properties of clomiphene: A review. Pharmacol. Ther. 1981, 15, 467–519. [Google Scholar] [CrossRef]
- Hart, J.E. Endocrine Pathology of Estrogens: Species Differnces. Pharmacol. Ther. 1990, 47, 203–218. [Google Scholar] [CrossRef]
- Sumida, K.; Ooe, N.; Saito, K.; Kaneko, H. Limited species differences in estrogen receptor alpha-medicated reporter gene transactivation by xenoestrogens. J. Steroid Biochem. Mol. Biol. 2003, 84, 33–40. [Google Scholar] [CrossRef]
- Petit, F.G.; Valotaire, Y.; Pakdel, F. The analysis of chimeric human/rainbow trout estrogen receptors reveals amino acid residues outside of P- and D-boxes important for the transactivation function. Nucleic Acids Res. 2000, 28, 2634–2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arao, Y.; Korach, K.S. The F domain of estrogen receptor α is involved in species-specific, tamoxifen-mediated transactivation. J. Biol. Chem. 2018, 293, 8495–8507. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.A.; Zhong, L.; Deighton-Collins, S.; Zhao, C.; Skafar, D.F. Mutations targeted to a predicted helix in the extreme carboxyl-terminal region of the human estrogen receptor-alpha alter its response to estradiol and 4-hydroxytamoxifen. J. Biol. Chem. 2002, 277, 13202–13209. [Google Scholar] [CrossRef] [PubMed]
- De Vries-van Leeuwen, I.J.; da Costa Pereira, D.; Flach, K.D.; Piersma, S.R.; Haase, C.; Bier, D.; Yalcin, Z.; Michalides, R.; Feenstra, K.A.; Jiménez, C.R.; et al. Interaction of 14-3-3 proteins with the estrogen receptor alpha F domain provides a drug target interface. Proc. Natl. Acad. Sci. USA 2013, 110, 8894–8899. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, M.; Ruff, M.; Eiler, S.; Duclaud, S.; Wurtz, J.M.; Moras, D. Crystal structure of a mutant hERalpha ligand-binding domain reveals key structural features for the mechanism of partial agonism. J. Biol. Chem. 2001, 276, 15059–15065. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-L.; Yang, X.; Ren, Z.; McDonnell, D.P.; Norris, J.D.; Willson, T.M.; Greene, G.L. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 2005, 18, 413–424. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arao, Y.; Korach, K.S. Transactivation Function-1-Mediated Partial Agonist Activity of Selective Estrogen Receptor Modulator Requires Homo-Dimerization of the Estrogen Receptor α Ligand Binding Domain. Int. J. Mol. Sci. 2019, 20, 3718. https://doi.org/10.3390/ijms20153718
Arao Y, Korach KS. Transactivation Function-1-Mediated Partial Agonist Activity of Selective Estrogen Receptor Modulator Requires Homo-Dimerization of the Estrogen Receptor α Ligand Binding Domain. International Journal of Molecular Sciences. 2019; 20(15):3718. https://doi.org/10.3390/ijms20153718
Chicago/Turabian StyleArao, Yukitomo, and Kenneth S. Korach. 2019. "Transactivation Function-1-Mediated Partial Agonist Activity of Selective Estrogen Receptor Modulator Requires Homo-Dimerization of the Estrogen Receptor α Ligand Binding Domain" International Journal of Molecular Sciences 20, no. 15: 3718. https://doi.org/10.3390/ijms20153718