Consistent Biomarkers and Related Pathogenesis Underlying Asthma Revealed by Systems Biology Approach

 ,
,
Abstract
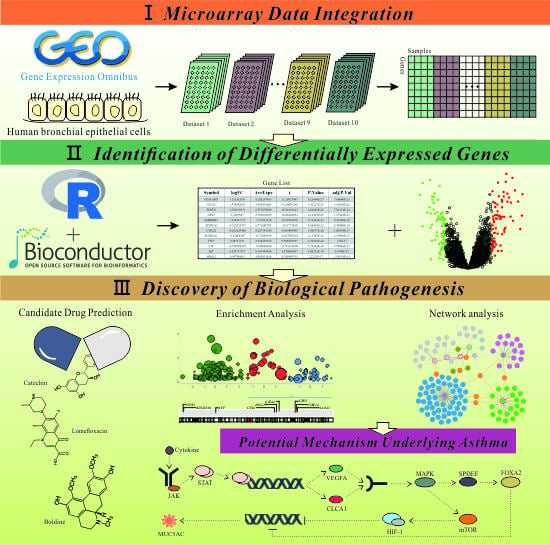
1. Introduction
2. Results and Discussion
2.1. Ten Independent Datasets Meeting the Criteria in This Study
2.2. Large Microarray Dataset Generated from Ten Selective Datasets
2.2.1. Sample Quality Control and Microarray Data Preprocessing
2.2.2. Data Integration and Batch Correction
2.3. Identification of DEGs Involved in Pathogenesis of Asthma
2.4. Functional Annotations and Enrichment Analysis of DEGs
2.4.1. Gene Ontology Analysis of DEGs
2.4.2. KEGG Pathway Analysis of DEGs
2.4.3. Potential Target Sites of Transcription Factors and Regulatory MicroRNAs
2.4.4. Effect of Chromosomal Position on the Expression of DEGs
2.5. Identification of Hub Genes Based on PPI Network Construction
2.6. Identification of Candidate Small Molecules
2.7. Crosstalk Pathway of Asthma
3. Materials and Methods
3.1. Microarray Gene Expression Data Acquisition
3.2. Quality Control and Individual Microarray Dataset Preprocessing
3.3. Data Integration and Batch Effect Removal
3.4. Identification of Differentially Expressed Genes
3.5. Gene Ontology and Pathway Enrichment Analysis
3.6. Protein-Protein Interaction Network Construction and Community Detection
3.7. Target Gene Prediction of Key Transcription Factors and Regulatory MicroRNAs
3.8. Chromosome Position Effect on Gene Expression
3.9. Connection of DEGs and Small Chemical Molecules
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DEGs | Differentially Expressed Genes |
| GSEA | Gene Set Enrichment Analysis |
| PPI | Protein-protein Interaction |
| CMap | Connectivity Map |
| ES | Enrichment Score |
References
- Willis-Owen, S.A.G.; Cookson, W.O.C.; Moffatt, M.F. The Genetics and Genomics of Asthma. Annu. Rev. Genomics Hum. Genet. 2018, 19, 223–246. [Google Scholar] [CrossRef] [PubMed]
- Lemmetyinen, R.; Karjalainen, J.; But, A.; Renkonen, R.; Pekkanen, J.; Toppila-Salmi, S.; Haukka, J. Higher mortality of adults with asthma: A 15-year follow-up of a population-based cohort. Allergy 2018, 73, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Dhami, S.; Kakourou, A.; Asamoah, F.; Agache, I.; Lau, S.; Jutel, M.; Muraro, A.; Roberts, G.; Akdis, C.A.; Bonini, M. Allergen immunotherapy for allergic asthma: A systematic review and meta-analysis. Allergy 2017, 72, 1825–1848. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Yang, T.; Xu, J.; Yang, L.; Zhao, J.; Zhang, X.; Bai, C.; Kang, J.; Ran, P.; Shen, H. Prevalence, risk factors, and management of asthma in China: A national cross-sectional study. Lancet 2019. [Google Scholar] [CrossRef]
- Forno, E.; Celedon, J.C. Epigenomics and Transcriptomics in the Prediction and Diagnosis of Childhood Asthma: Are We There Yet? Front. Pediatr. 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Zhao, W.; Lin, Y.; Yao, L.; Huang, G.; Yu, C.; Dong, H.; Xiao, G.; Zhao, H.; Cai, S. Phosphorylation of low density lipoprotein receptor-related protein 6 is involved in receptor for advanced glycation end product-mediated β-catenin stabilization in a toluene diisocyanate-induced asthma model. Int. Immunopharmacol. 2018, 59, 187–196. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Boushey, H.A.; Dolganov, G.M.; Barker, C.S.; Yang, Y.H.; Donnelly, S.; Ellwanger, A.; Sidhu, S.S.; Dao-Pick, T.P.; Pantoja, C.; et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc. Natl. Acad. Sci. USA 2007, 104, 15858–15863. [Google Scholar] [CrossRef]
- Kicic, A.; Hallstrand, T.S.; Sutanto, E.N.; Stevens, P.T.; Kobor, M.S.; Taplin, C.; Pare, P.D.; Beyer, R.P.; Stick, S.M.; Knight, D.A. Decreased fibronectin production significantly contributes to dysregulated repair of asthmatic epithelium. Am. J. Respir. Crit. Care Med. 2010, 181, 889–898. [Google Scholar] [CrossRef]
- Modena, B.D.; Tedrow, J.R.; Milosevic, J.; Bleecker, E.R.; Meyers, D.A.; Wu, W.; Bar-Joseph, Z.; Erzurum, S.C.; Gaston, B.M.; Busse, W.W.; et al. Gene expression in relation to exhaled nitric oxide identifies novel asthma phenotypes with unique biomolecular pathways. Am. J. Respir. Crit. Care Med. 2014, 190, 1363–1372. [Google Scholar] [CrossRef]
- Singhania, A.; Wallington, J.C.; Smith, C.G.; Horowitz, D.; Staples, K.J.; Howarth, P.H.; Gadola, S.D.; Djukanović, R.; Woelk, C.H.; Hinks, T.S. Multitissue transcriptomics delineates the diversity of airway T cell functions in asthma. Am. J. Respir. Cell Mol. Biol. 2018, 58, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299. [Google Scholar] [CrossRef] [PubMed]
- Rung, J.; Brazma, A. Reuse of public genome-wide gene expression data. Nat. Rev. Genet. 2013, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Wagener, A.H.; Zwinderman, A.H.; Luiten, S.; Fokkens, W.J.; Bel, E.H.; Sterk, P.J.; van Drunen, C.M. The impact of allergic rhinitis and asthma on human nasal and bronchial epithelial gene expression. PLoS ONE 2013, 8, e80257. [Google Scholar] [CrossRef] [PubMed]
- Singhania, A.; Rupani, H.; Jayasekera, N.; Lumb, S.; Hales, P.; Gozzard, N.; Davies, D.E.; Woelk, C.H.; Howarth, P.H. Altered Epithelial Gene Expression in Peripheral Airways of Severe Asthma. PLoS ONE 2017, 12, e0168680. [Google Scholar] [CrossRef] [PubMed]
- Christenson, S.A.; Steiling, K.; van den Berge, M.; Hijazi, K.; Hiemstra, P.S.; Postma, D.S.; Lenburg, M.E.; Spira, A.; Woodruff, P.G. Asthma-COPD overlap. Clinical relevance of genomic signatures of type 2 inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2015, 191, 758–766. [Google Scholar] [CrossRef]
- Yang, I.V.; Richards, A.; Davidson, E.J.; Stevens, A.D.; Kolakowski, C.A.; Martin, R.J.; Schwartz, D.A. The Nasal Methylome: A Key to Understanding Allergic Asthma. Am. J. Respir. Crit. Care Med. 2017, 195, 829–831. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Greer, E.L.; Brunet, A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef]
- Michnick, S.W. The connectivity map. Nat. Chem. Biol. 2006, 2, 663–664. [Google Scholar] [CrossRef]
- Brooks, W.H.; Renaudineau, Y. Epigenetics and autoimmune diseases: The X chromosome-nucleolus nexus. Front. Genet. 2015, 6, 22. [Google Scholar] [CrossRef]
- Rava, M.; Czachorowski, M.J.; Silverman, D.; Márquez, M.; Kishore, S.; Tardón, A.; Serra, C.; García-Closas, M.; Garcia-Closas, R.; Carrato, A. Asthma status is associated with decreased risk of aggressive urothelial bladder cancer. Int. J. Cancer. 2018, 142, 470–476. [Google Scholar] [CrossRef]
- Fish, J.E.; Ankin, M.G.; Adkinson, N.F., Jr.; Peterman, V.I. Indomethacin modification of immediate-type immunologic airway responses in allergic asthmatic and non-asthmatic subjects: Evidence for altered arachidonic acid metabolism in asthma. Am. Rev. Respir. Dis. 1981, 123, 609–614. [Google Scholar] [PubMed]
- Cottrell, L.; Neal, W.A.; Ice, C.; Perez, M.K.; Piedimonte, G. Metabolic abnormalities in children with asthma. Am. J. Respir. Crit. Care Med. 2011, 183, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Woods, R.; Raven, J.; Walters, E.; Abramson, M.; Thien, F. Fatty acid levels and risk of asthma in young adults. Thorax 2004, 59, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Devereux, G.; Seaton, A. Diet as a risk factor for atopy and asthma. J. Allergy Clin. Immunol. 2005, 115, 1109–1117. [Google Scholar] [CrossRef] [PubMed]
- Mahn, K.; Ojo, O.O.; Chadwick, G.; Aaronson, P.I.; Ward, J.P.; Lee, T.H. Ca2+ homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax 2010, 65, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Fritscher, L.; Chapman, K.R. Seretide: A pharmacoeconomic analysis. J. Med. Econ. 2008, 11, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.-h.; Ding, Z.; Narurkar, R.; Ramkissoon, S.; Muller, F.; Kamoun, W.S.; Chae, S.-S.; Zheng, H.; Ying, H.; Mahoney, J. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell stem cell 2009, 5, 540–553. [Google Scholar] [CrossRef]
- Schroeder, B.W.; Verhaeghe, C.; Park, S.-W.; Nguyenvu, L.T.; Huang, X.; Zhen, G.; Erle, D.J. AGR2 is induced in asthma and promotes allergen-induced mucin overproduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 178–185. [Google Scholar] [CrossRef]
- Shi, N.; Zhang, J.; Chen, S.-Y. Runx2, a novel regulator for goblet cell differentiation and asthma development. FASEB J. 2016, 31, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Makrinioti, H.; Toussaint, M.; Jackson, D.J.; Walton, R.P.; Johnston, S.L. Role of interleukin 33 in respiratory allergy and asthma. Lancet Respir. Med. 2014, 2, 226–237. [Google Scholar] [CrossRef]
- Zhou, X.; Kinlough, C.L.; Hughey, R.P.; Jin, M.; Inoue, H.; Etling, E.; Modena, B.D.; Kaminski, N.; Bleecker, E.R.; Meyers, D.A. Sialylation of MUC4β N-glycans by ST6GAL1 orchestrates human airway epithelial cell differentiation associated with type-2 inflammation. JCI insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T.; Holloway, J.; Wilson, S.; Bucchieri, F.; Puddicombe, S.; Davies, D.E. Epithelial–mesenchymal communication in the pathogenesis of chronic asthma. Proc. Am. Thorac. Soc. 2004, 1, 93–98. [Google Scholar] [CrossRef]
- Feng, M.-J.; Shi, F.; Qiu, C.; Peng, W.-K. MicroRNA-181a,-146a and-146b in spleen CD4+ T lymphocytes play proinflammatory roles in a murine model of asthma. Int. Immunopharmacol. 2012, 13, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, J.S.; Lopez, M.A.; Boriek, A.M. Mechanical stretch up-regulates microRNA-26a and induces human airway smooth muscle hypertrophy by suppressing glycogen synthase kinase-3β. J. Biol. Chem. 2010, 285, 29336–29347. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.; Jacobs, K.B.; Yeager, M.; Kraft, P.; Wacholder, S.; Orr, N.; Yu, K.; Chatterjee, N.; Welch, R.; Hutchinson, A. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 2008, 40, 310. [Google Scholar] [CrossRef]
- Thomas, G.; Jacobs, K.B.; Kraft, P.; Yeager, M.; Wacholder, S.; Cox, D.G.; Hankinson, S.E.; Hutchinson, A.; Wang, Z.; Yu, K. A multistage genome-wide association study in breast cancer identifies two new risk alleles at 1p11. 2 and 14q24. 1 (RAD51L1). Nat. Genet. 2009, 41, 579. [Google Scholar] [CrossRef]
- Celedon, J.C.; Soto-Quiros, M.E.; Avila, L.; Lake, S.L.; Liang, C.; Fournier, E.; Spesny, M.; Hersh, C.P.; Sylvia, J.S.; Hudson, T.J.; et al. Significant linkage to airway responsiveness on chromosome 12q24 in families of children with asthma in Costa Rica. Hum. Genet. 2007, 120, 691–699. [Google Scholar] [CrossRef]
- Ferreira, M.A.; Matheson, M.C.; Duffy, D.L.; Marks, G.B.; Hui, J.; Le Souëf, P.; Danoy, P.; Baltic, S.; Nyholt, D.R.; Jenkins, M. Identification of IL6R and chromosome 11q13. 5 as risk loci for asthma. Lancet 2011, 378, 1006–1014. [Google Scholar] [CrossRef]
- Zhou, G.; Soufan, O.; Ewald, J.; Hancock, R.E.; Basu, N.; Xia, J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Peroni, D.G.; Djukanovic, R.; Bradding, P.; Feather, I.H.; Montefort, S.; Howarth, P.H.; Jones, D.B.; Holgate, S.T. Expression of CD44 and integrins in bronchial mucosa of normal and mildly asthmatic subjects. Eur. Respir. J. 1996, 9, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Sano, K.; Yamauchi, K.; Hoshi, H.; Honma, M.; Tamura, G.; Shirato, K. CD44 expression on blood eosinophils is a novel marker of bronchial asthma. Int. Arch. Allergy Immunol. 1997, 114, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Yang, S.; Zhang, Z.; Cui, Y.; Quan, C.; Zhou, F.; Fang, Q.; Du, W.; Zhang, F.; Chang, J. Novel and recurrent keratin 6A (KRT6A) mutations in Chinese patients with pachyonychia congenita type 1. Br. J. Dermatol. 2009, 160, 1327–1329. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, L.-C.; Song, X.; Lu, J.-R.; Jin, Z. KRT6 interacting with notch1 contributes to progression of renal cell carcinoma, and aliskiren inhibits renal carcinoma cell lines proliferation in vitro. Int. J. Clin. Exp. Pathol. 2015, 8, 9182. [Google Scholar] [PubMed]
- Bu, W.; Chen, J.; Morrison, G.D.; Huang, S.; Creighton, C.J.; Huang, J.; Chamness, G.C.; Hilsenbeck, S.G.; Roop, D.R.; Leavitt, A.D. Keratin 6a marks mammary bipotential progenitor cells that can give rise to a unique tumor model resembling human normal-like breast cancer. Oncogene 2011, 30, 4399. [Google Scholar] [CrossRef]
- Gao, P.; Grigoryev, D.; Breslin, L.; Cheadle, C.; Mathias, R.; Beaty, T.; Togias, A.; Barnes, K. Keratins: Important candidate genes for asthma and immune responsiveness to cockroach. J. Allergy Clin. Immunol. 2007, 119, S176. [Google Scholar] [CrossRef]
- Zhang, C.; Abudula, A.; Awuti, M.; Wang, H.; Aihemaiti, X.; Tusung, T.; Sulaiman, X.; Upur, H. Plasma proteins as potential targets of abnormal Savda syndrome in asthma patients treated with unique Uighur prescription. Exp. Ther. Med. 2017, 14, 267–275. [Google Scholar] [CrossRef]
- Fang, F.; Pan, J.; Li, Y.; Li, Y.; Feng, X.; Wang, J. Identification of potential transcriptomic markers in developing asthma: An integrative analysis of gene expression profiles. Mol. Immunol. 2017, 92, 38–44. [Google Scholar] [CrossRef]
- Kruzel, M.L.; Bacsi, A.; Choudhury, B.; Sur, S.; Boldogh, I. Lactoferrin decreases pollen antigen-induced allergic airway inflammation in a murine model of asthma. Immunology 2006, 119, 159–166. [Google Scholar] [CrossRef]
- Tanner, A.; Ward, J.; Wilson, S.; Howarth, P. S30 CEACAM5 (CD66e) mucosal immunoreactivity and its relationship to asthma. Thorax 2018, 73, A18. [Google Scholar]
- Adcock, I.M.; Loza, M.; Djukanovic, R.; Rowe, A.; Rao, N.; Chung, K.F.; Baribaud, F. Differential gene expression profiles of endobronchial biopsies from severe and non-severe asthmatics obtained in UBIOPRED. Am. J. Respir. Crit. Care Med. 2014, A2422. [Google Scholar]
- de Jong, E.; Bosco, A. Dissecting Asthma Transcriptomics: Does Site Matter? Am. J. Respir. Cell Mol. Biol. 2018, 58, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Naumov, D.; Gassan, D.; Kotova, O.; Prikhodko, A.; Pinegina, E.; Perelman, J.; Kolosov, V.; Zhou, X.; Li, Q. Cold air alters MUC5AC and MUC5B expression in the airways of asthma patients. Eur. Respir. J. 2018, 52 (Suppl. 62), PA1272. [Google Scholar]
- Shrine, N.; Portelli, M.A.; John, C.; Artigas, M.S.; Bennett, N.; Hall, R.; Lewis, J.; Henry, A.P.; Billington, C.K.; Ahmad, A. Moderate-to-severe asthma in individuals of European ancestry: A genome-wide association study. Lancet Respir. Med. 2019, 7, 20–34. [Google Scholar] [CrossRef]
- Zaki, N.; Mora, A. A comparative analysis of computational approaches and algorithms for protein subcomplex identification. Sci. Rep. 2014, 4, 4262. [Google Scholar] [CrossRef] [PubMed]
- Nagata, H.; Takekoshi, S.; Takagi, T.; Honma, T.; Watanabe, K. Antioxidative action of flavonoids, quercetin and catechin, mediated by the activation of glutathione peroxidase. Tokai J. Exp. Clin. Med. 1999, 24, 1–11. [Google Scholar]
- Yamane, T.; Nakatani, H.; Kikuoka, N.; Matsumoto, H.; Iwata, Y.; Kitao, Y.; Oya, K.; Takahashi, T. Inhibitory effects and toxicity of green tea polyphenols for gastrointestinal carcinogenesis. Cancer 1996, 77, 1662–1667. [Google Scholar] [CrossRef]
- Fujimura, Y.; Tachibana, H.; Maeda-Yamamoto, M.; Miyase, T.; Sano, M.; Yamada, K. Antiallergic tea catechin,(−)-epigallocatechin-3-O-(3-O-methyl)-gallate, suppresses FcεRI expression in human basophilic KU812 cells. J. Agric. Food Chem. 2002, 50, 5729–5734. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, H.J.; Lee, C.M.; Choi, I.W.; Moon, D.O.; Roh, H.J.; Lee, H.K.; Park, Y.M. Epigallocatechin-3-gallate protects toluene diisocyanate-induced airway inflammation in a murine model of asthma. FEBS Lett. 2006, 580, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Patel, V. Inhibitory effects of catechin isolated from Acacia catechu on ovalbumin induced allergic asthma model: Role of histidine decarboxylase. Nutr. & Food Sci. 2019, 49, 18–31. [Google Scholar]
- Grassi, C.; Albera, C.; Pozzi, E. Lomefloxacin versus amoxicillin in the treatment of acute exacerbations of chronic bronchitis: An Italian multicenter study. Am. J. Med. 1992, 92, S103–S107. [Google Scholar] [CrossRef]
- Todd, I.; Negm, O.H.; Reps, J.; Radford, P.; Figueredo, G.; McDermott, E.M.; Drewe, E.; Powell, R.J.; Bainbridge, S.; Hamed, M. A signalome screening approach in the autoinflammatory disease TNF receptor associated periodic syndrome (TRAPS) highlights the anti-inflammatory properties of drugs for repurposing. Pharmacol. Res. 2017, 125, 188–200. [Google Scholar] [CrossRef]
- de Lima, N.M.R.; Ferreira, E.d.O.; Fernandes, M.Y.S.; Lima, F.A.V.; Neves, K.R.T.; do Carmo, M.R.S.; de Andrade, G.M. Neuroinflammatory response to experimental stroke is inhibited by boldine. Behav. pharmacol. 2017, 28, 223–237. [Google Scholar] [CrossRef]
- Fernández, J.; Lagos, P.; Rivera, P.; Zamorano-Ponce, E. Effect of boldo (Peumus boldus Molina) infusion on lipoperoxidation induced by cisplatin in mice liver. Phytother. Res. 2009, 23, 1024–1027. [Google Scholar] [CrossRef]
- Liu, G.; Cooley, M.A.; Nair, P.M.; Donovan, C.; Hsu, A.C.; Jarnicki, A.G.; Haw, T.J.; Hansbro, N.G.; Ge, Q.; Brown, A.C. Airway remodelling and inflammation in asthma are dependent on the extracellular matrix protein fibulin-1c. J. Pathol. 2017, 243, 510–523. [Google Scholar] [CrossRef]
- Agache, I.; Akdis, C.; Jutel, M.; Virchow, J. Untangling asthma phenotypes and endotypes. Allergy 2012, 67, 835–846. [Google Scholar] [CrossRef]
- Ma, L.; Javier, C.; Al Kolla, R.; Moshensky, A.; Bojanowski, C.; Shin, J.; Crotty Alexander, L.; Chun, V. HIFα Subunits Alter Asthma Phenotype in a Murine Model. Am. J. Respir. Crit. Care Med. 2018, 197, A1332. [Google Scholar]
- Dewitz, C.; McEachern, E.; Shin, S.; Akong, K.; Nagle, D.G.; Broide, D.H.; Akuthota, P.; Alexander, L.E.C. Hypoxia-inducible factor-1α inhibition modulates airway hyperresponsiveness and nitric oxide levels in a BALB/c mouse model of asthma. Clin. Immunol. 2017, 176, 94–99. [Google Scholar] [CrossRef]
- Ahmad, T.; Kumar, M.; Mabalirajan, U.; Pattnaik, B.; Aggarwal, S.; Singh, R.; Singh, S.; Mukerji, M.; Ghosh, B.; Agrawal, A. Hypoxia response in asthma: Differential modulation on inflammation and epithelial injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 1–10. [Google Scholar] [CrossRef]
- Wu, S.; Li, H.; Yu, L.; Wang, N.; Li, X.; Chen, W. IL-1β upregulates Muc5ac expression via NF-κB-induced HIF-1α in asthma. Immunol. lett. 2017, 192, 20–26. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann, A.; Gentleman, R.; Huber, W. arrayQualityMetrics--a bioconductor package for quality assessment of microarray data. Bioinformatics 2009, 25, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Irizarry, R.A. Preprocessing of oligonucleotide array data. Nat. Biotechnol. 2004, 22, 656–658, 656–658; author reply 658. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef]
- Letellier, E.; Schmitz, M.; Ginolhac, A.; Rodriguez, F.; Ullmann, P.; Qureshi-Baig, K.; Frasquilho, S.; Antunes, L.; Haan, S. Loss of Myosin Vb in colorectal cancer is a strong prognostic factor for disease recurrence. Br. J. Cancer 2017, 117, 1689. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, Y.; Zhang, S.; Tang, J.; Li, F.; Yin, J.; Li, Y.; Fu, J.; Li, B.; Luo, Y. Biomarker discovery for immunotherapy of pituitary adenomas: Enhanced robustness and prediction ability by modern computational tools. Int. J. Mol. Sci. 2019, 20, 151. [Google Scholar] [CrossRef]
- Leek, J.T.; Johnson, W.E.; Parker, H.S.; Jaffe, A.E.; Storey, J.D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 2012, 28, 882–883. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Walter, W.; Sánchez-Cabo, F.; Ricote, M. GOplot: An R package for visually combining expression data with functional analysis. Bioinformatics 2015, 31, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C. Pathway enrichment analysis and visualization of omics data using g: Profiler, GSEA, Cytoscape and EnrichmentMap. Nat. Protoc. 2019, 14, 482. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009, 37, W305–311. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007, 2, 2366. [Google Scholar] [CrossRef] [PubMed]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, P.; Safikhani, Z.; El-Hachem, N.; Wang, D.; She, A.; Olsen, C.; Freeman, M.; Selby, H.; Gendoo, D.M.; Grossmann, P.; et al. PharmacoGx: An R package for analysis of large pharmacogenomic datasets. Bioinformatics 2016, 32, 1244–1246. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO ID | Microarray Platform | Sample Size (A/H) 1 | Cell Type | Author (Year) |
|---|---|---|---|---|
| GSE470 | HG-U95Av2 | 12 (6/6) | Epithelial cells | Spannhake W, et al. (2003) |
| GSE4302 | HG-U133_Plus_2 | 70 (42/28) | Airway epithelial cells | Woodruff PG, et al. (2007) [8] |
| GSE18965 | HG-U133A | 16 (9/7) | Bronchial epithelial cells | Beyer RP, et al. (2010) [9] |
| GSE41861 | HG-U133_Plus_2 | 81 (51/30) | Bronchial epithelial cells | Cheng DT, et al. (2015) |
| GSE44037 | HT_HG-U133_Plus_PM | 12 (6/6) | Bronchial epithelia | Wagener AH, et al. (2013) [14] |
| GSE63142 | GPL6480 (Agilent) | 155 (128/27) | Bronchial epithelia | Wenzel S, et al. (2014) [10] |
| GSE64913 | HG-U133_Plus_2 | 59 (22/37) | Peripheral airway epithelia | Singhania A, et al. (2017) [15] |
| GSE67472 | HG-U133_Plus_2 | 105 (62/43) | Airway epithelia | Christenson SA, et al. (2015) [16] |
| GSE89809 | HT_HG-U133_Plus_PM | 56 (38/18) | Epithelial cells | Singhania A, et al. (2017) [11] |
| GSE104468 | GPL21185 (Agilent) | 24 (12/12) | Bronchial epithelia | Richards A, et al. (2017) [17] |
| Gene | Entrez ID | Log2(Fold Change) | Asthmatics vs. Healthy Controls | FDR 1 |
|---|---|---|---|---|
| CEACAM5 | 1048 | 1.13 | Up | 7.67 × 10−23 |
| CLCA1 | 1179 | 1.58 | Up | 5.43 × 10−22 |
| POSTN | 10631 | 1.33 | Up | 7.83 × 10−22 |
| CPA3 | 1359 | 1.26 | Up | 1.28 × 10−21 |
| SERPINB2 | 5055 | 1.14 | Up | 9.55 × 10−20 |
| LTF | 4057 | −0.75 | Down | 4.60 × 10−17 |
| MUC5B | 727897 | −0.89 | Down | 2.10 × 10−13 |
| KRT6A | 3853 | 0.61 | Up | 4.89 × 10−12 |
| CD44 | 960 | 0.60 | Up | 9.03 × 10−9 |
| MUC5AC | 4586 | 0.59 | Up | 1.03 × 10−6 |
| GO Term | Description | Count 1 | Z-score | Adj. p 2 | Category |
|---|---|---|---|---|---|
| GO:0042221 | Response to chemical stimulus | 34 | −0.34 | 2.64 × 10−4 | BP |
| GO:0032501 | Multicellular organismal process | 69 | 0.12 | 1.74 × 10−3 | BP |
| GO:0018149 | Peptide cross-linking | 6 | 0.00 | 2.50 × 10−3 | BP |
| GO:0005576 | Extracellular region | 51 | 0.42 | 1.67 × 10−8 | CC |
| GO:0044421 | Extracellular region part | 31 | 0.18 | 4.21 × 10−7 | CC |
| GO:0005615 | Extracellular space | 21 | −0.65 | 4.33 × 10−4 | CC |
| GO:0030141 | Secretory granule | 10 | 0.63 | 2.75 × 10−3 | CC |
| GO:0031012 | Extracellular matrix | 13 | 0.28 | 4.33 × 10−3 | CC |
| GO:0000267 | Cell fraction | 24 | −0.82 | 6.39 × 10−3 | CC |
| GO:0005624 | Membrane fraction | 20 | −0.89 | 6.10 × 10−3 | CC |
| GO:0005578 | Proteinaceous extracellular matrix | 12 | 0.00 | 5.90 × 10−3 | CC |
| GO:0005626 | Insoluble fraction | 20 | −0.89 | 7.52 × 10−3 | CC |
| KEGG Pathway | FDR 1 | Expression Pattern 2 | Z-score | Gene |
|---|---|---|---|---|
| (1) Pathways in cancer | 3.24 × 10−5 | 3↑+ 7↓ | −1.26 | NOS2, MMP1, VEGFA, DAPK1, FOS, KIT, FN1, RUNX1T1, EPAS1, AR |
| (2) Arachidonic acid metabolism | 1.42 × 10−4 | 3↑+ 2↓ | 0.45 | CYP2J2, ALOX15, GPX3, HPGDS, PTGS1 |
| (3) Linoleic acid metabolism | 7.69 × 10−3 | 2↑+ 1↓ | 0.58 | CYP2J2, ALOX15, AKR1B10 |
| (4) Calcium signaling pathway | 1.17 × 10−2 | 1↑+ 4↓ | −1.34 | NOS2, ITPR1, AVPR1A, PTGFR, TRPC1 |
| (5) Aldosterone-regulated sodium reabsorption | 1.17 × 10−2 | 0↑+ 3↓ | −1.73 | IRS2, INSR, SCNN1G |
| (6) Bladder cancer | 1.17 × 10−2 | 1↑+ 2↓ | −0.58 | MMP1, VEGFA, DAPK1 |
| (7) Arginine and proline metabolism | 1.95 × 10−2 | 2↑+ 1↓ | 0.58 | NOS2, ODC1, PYCR1 |
| (8) PPAR signaling pathway | 3.34 × 10−2 | 2↑+ 1↓ | 0.58 | MMP1, CD36, FABP6 |
| (9) Leishmania infection | 3.39 × 10−2 | 1↑+ 2↓ | −0.58 | NOS2, FOS, HLA-DQB1 |
| (10) Cytokine-cytokine receptor interaction | 3.51 × 10−2 | 2↑+ 3↓ | −0.45 | VEGFA, KIT, CXCL2, CXCL6, CSF2RB |
| (11) ECM-receptor interaction | 4.39 × 10−2 | 2↑+ 1↓ | 0.58 | FN1, CD36, CD44 |
| (12) Hematopoietic cell lineage | 4.62 × 10−2 | 3↑+ 0↓ | 1.73 | KIT, CD36, CD44 |
| Molecules | Enrichment Score | p-Value |
|---|---|---|
| Catechin | −0.7995 | 0.0066 |
| Lomefloxacin | −0.7101 | 0.0072 |
| Boldine | −0.6635 | 0.0250 |
| Prestwick-1082 | 0.6944 | 0.0099 |
| Ricinine | 0.7297 | 0.0102 |
| Milrinone | 0.7751 | 0.0153 |
| Econazole | 0.7981 | 0.0170 |
| Acetohexamide | 0.6542 | 0.0181 |
| Cefsulodin | 0.7900 | 0.0192 |
| Nifuroxazide | 0.7567 | 0.0199 |
| Alimemazine | 0.7744 | 0.0289 |
| Progesterone | 0.7258 | 0.0309 |
| Zoxazolamine | 0.7206 | 0.0390 |
| Colistin | 0.6503 | 0.0404 |
| Methapyrilene | 0.7618 | 0.0427 |
| Tiapride | 0.6832 | 0.0436 |
| Fluocinonide | 0.7451 | 0.0466 |
| Ganciclovir | 0.7389 | 0.0466 |
| Quinisocaine | 0.6790 | 0.0479 |
| Mexiletine | 0.7615 | 0.0492 |
| Pathway ID | Name | Database | FDR | Count 1 |
|---|---|---|---|---|
| M5889 | Genes encoding ECM and ECM-associated proteins | BIOCARTA (v6.0) | 1.40 × 10−2 | 26 |
| M5885 | ECM-affiliated proteins, regulators and secreted factors | BIOCARTA (v6.0) | 2.42 × 10−2 | 20 |
| 1470923 | Interleukin−4 and 13 signaling | REACTOME | 1.40 × 10−2 | 8 |
| 138045 | HIF−1-alpha transcription factor network | Pathway Interaction Database | 2.15 × 10−2 | 6 |
| 137979 | FOXA1 transcription factor network | Pathway Interaction Database | 2.23 × 10−2 | 5 |
| 1268737 | Termination of O-glycan biosynthesis | REACTOME | 2.61 × 10−2 | 4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nie, X.; Wei, J.; Hao, Y.; Tao, J.; Li, Y.; Liu, M.; Xu, B.; Li, B. Consistent Biomarkers and Related Pathogenesis Underlying Asthma Revealed by Systems Biology Approach. Int. J. Mol. Sci. 2019, 20, 4037. https://doi.org/10.3390/ijms20164037
Nie X, Wei J, Hao Y, Tao J, Li Y, Liu M, Xu B, Li B. Consistent Biomarkers and Related Pathogenesis Underlying Asthma Revealed by Systems Biology Approach. International Journal of Molecular Sciences. 2019; 20(16):4037. https://doi.org/10.3390/ijms20164037
Chicago/Turabian StyleNie, Xiner, Jinyi Wei, Youjin Hao, Jingxin Tao, Yinghong Li, Mingwei Liu, Boying Xu, and Bo Li. 2019. "Consistent Biomarkers and Related Pathogenesis Underlying Asthma Revealed by Systems Biology Approach" International Journal of Molecular Sciences 20, no. 16: 4037. https://doi.org/10.3390/ijms20164037
APA StyleNie, X., Wei, J., Hao, Y., Tao, J., Li, Y., Liu, M., Xu, B., & Li, B. (2019). Consistent Biomarkers and Related Pathogenesis Underlying Asthma Revealed by Systems Biology Approach. International Journal of Molecular Sciences, 20(16), 4037. https://doi.org/10.3390/ijms20164037