2.1. Docking Results and Binding Mode
In order to verify the reliability of the CDOCKER docking results, BMS22 and 47 inhibitors were docked to the active sites of TβR1 under the same docking conditions. The docking results show that the conformation of BMS22 obtained by CDOCKER docking was highly consistent with that of crystal, and the minimum root mean square deviation (RMSD) of heavy atoms was 0.2879, which indicates that the docking method is feasible and the docking results are credible. The active conformations of 47 molecules were obtained by the screening criteria of a higher docking score and similar binding mode with BMS22 in the crystal structure. Their conformations of ligands in the binding pocket of TβR1 are shown in
Figure 2Inhibitors usually bind to target proteins by electrostatic, hydrogen-bonding and hydrophobic interactions with amino acid residues around the active site. The receptor surface schematic diagram is shown in
Figure 2C and
Figure 3A. As shown in
Figure 3A, residue SER280 protrudes into the cavity of the receptor site and forms a Y-type binding pocket with residue SER280 as a convex point. Therefore, most TβR1 inhibitors are Y-shaped, which can coincide with the Y-type binding pocket in the receptor. Though there is no interaction between residue SER280 and the ligands in terms of crystal structure and the binding mode obtained by molecular docking, residue SER280 is an important structural feature of the binding pocket.
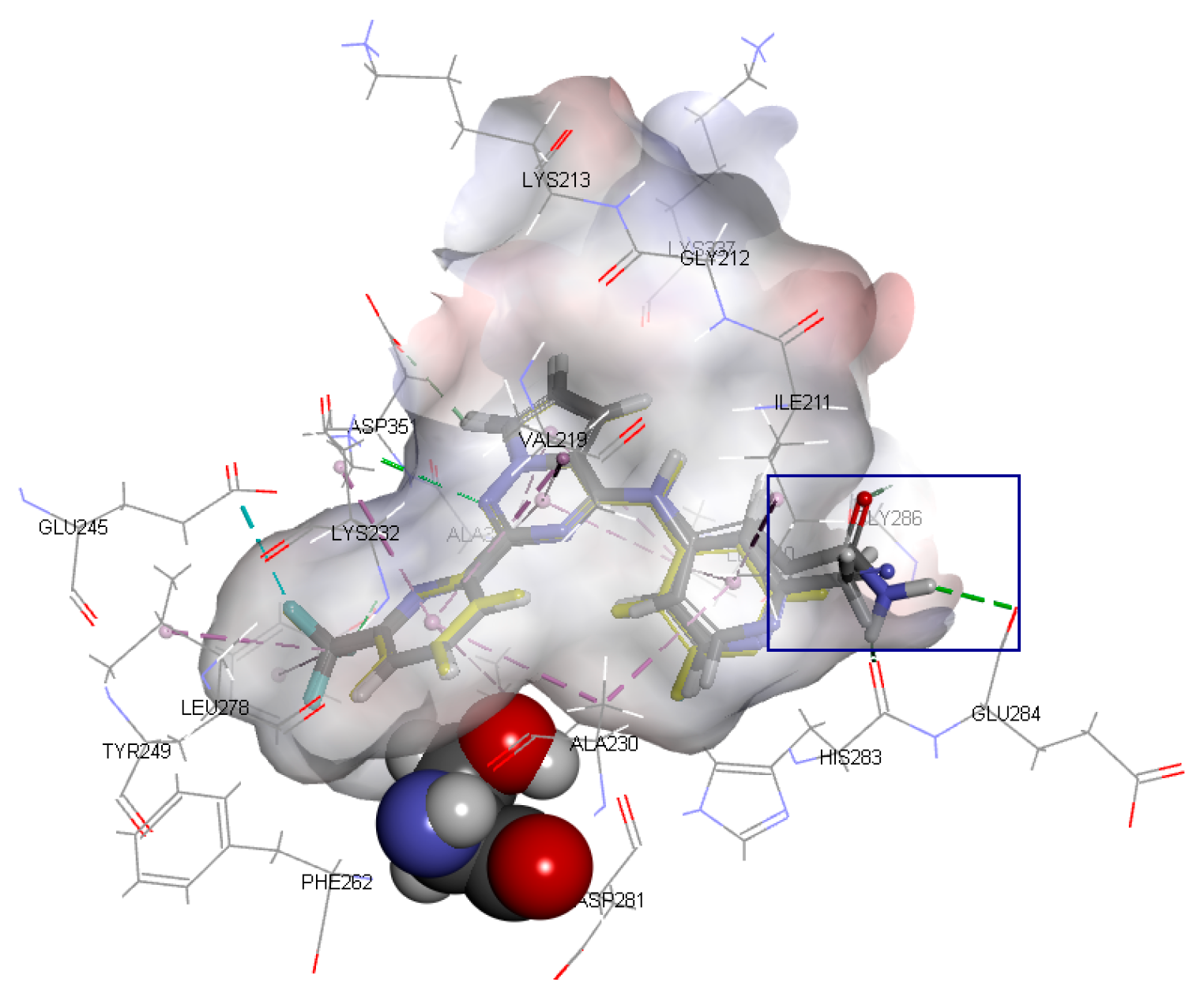
The binding mode of Compound 1 with the highest activity is shown in
Figure 3. As illustrated in
Figure 3A, amino acid residues such as ILE211, ALA230, and LEU340 form a cavity, and Ring A is inserted into it to form a stable spatial structure through hydrophobic interaction. Amino acid residues such as LYS232, LEU260 and LEU340 form a hydrophobic cavity, and Ring B extends into the cavity to form another stable spatial structure through hydrophobic interaction. In addition, a stable hydrophobic interaction was formed between Ring C and residues VAL219, LEU340, and ALA350. Aside from the hydrophobic effects, two important hydrogen bonds were formed between the ligand and the certain amino acid residues. The N1 atom on Ring A and residue HIS283 form an N···H-N hydrogen bond, while the N5 atom on Ring C and LYS232 form another N···H-N hydrogen bond.
On the basis of the above analysis, there are two characteristics of the binding mode between ligands and TβR1: (1) The hydrophobic interactions between three aromatic ring regions and amino acid residues are the key factors in the binding process; (2) the docking conformations of most active molecules indicate that the hydrogen bonds formed between the N1 and N5 atoms and the amino acid residues may be the crucial sites for pharmacological activity. On the premise of retaining the hydrogen bonds between the N1 and N5 atoms and the amino acid residues, new inhibitors may be found by modifying the molecular skeleton.
2.2. Structure—Activity Relationships
In order to investigate the substitution effect in the R1-position of Ring A, the docking conformation, molecular structure, and activity of the molecules with different substituents were compared and analyzed (
Figure 4 and
Table 1). As listed in
Table 1, the activity decreased significantly when the H atom in the R1-position of Compound 2 was substituted by other substituents. The introduction of -CH
3, -CONH
2, and -CN groups resulted in steric hindrance, which increased the space distance between the N1 atom and residue HIS283, and may have led to the disappearance or weakening of the N···H-N hydrogen bond, resulting in a decrease in activity. For example, when the hydrogen bonds between the N1 atoms and residues HIS283 in Compounds 3 and 4 disappeared, the pIC
50 (logIC
50) values reduce by 2.1474 and 2.2773, respectively, these values both being lower than Compound 22. The hydrogen bond length between the N1 atom in Compound 5 and residue HIS283 increased by 0.11 Å, then the hydrogen bond interactions were weakened, and the pIC
50 value decreased by 1.8842. The position of R1 was close to the electronegativity region of the receptor surface formed by amino acid residue HIS283, and the electronegativity groups in the R1-position, such as -F (Compound 6), may have caused electrostatic repulsion and decreased the activity of the receptor surface.
In conclusion, the steric hindrance or electrostatic repulsion caused by the substituent in the R1-position may have significantly reduced the activity, which was a disadvantageous site for the structural modification of compounds.
If there are small hydrophobic groups such as -Cl and -F in the R2-position of Ring A, the activity of the compound may significantly increase (data listed in
Table 2). The pIC50 value increased by 1.5550 or 1.2485 when -H in the R2-position of Compound 7 was replaced by -Cl (Compound 8) or -F (Compound 9). In addition, the values of pIC
50 increased by 0.1930 and 0.2218, respectively, after substituting -H in the R2-position of Compound 10 for -Cl (Compound 11) and -F (Compound 12). The value of pIC
50 increased by 0.2076 after replacing -H in the R2-position of Compound 13 with -F (Compound 14). The analysis of the binding mode (
Figure 5) illustrated that the halogen atoms in the R2-position can enhance the hydrophobic interaction with amino acid residues, which is conducive to ligand–receptor binding. Furthermore, the steric hindrance caused by the substituent in the R2-position could decrease the activity. For example, Compounds 15, 16, and 17 showed lower activity.
Considering all of these points, the conclusion can be drawn that the existence of small electronegative groups in the R2-position of cyclic A is beneficial in terms of increasing the activity, and the introduction of other groups should be considered in combination with steric hindrance and other factors.
As shown in
Figure 3, Ring B lied deep in the cavity of the receptor and was surrounded by hydrophobic amino acids. The hydrophobic group on Ring B was beneficial to increase the activity of the compounds; nevertheless, the activity decreased as a result of the polar group or steric hindrance caused by the substituent exists (
Table 3). For example, the pIC
50 value of Compound 9 increased by 0.2785 compared to Compound 12.
On the basis of the characteristics of the cavity structure, the steric hindrance caused by the substituent groups in the R3-position of Ring B was a disadvantage factor; meanwhile, the small hydrophobic groups in the R3-position may have increased hydrophobic interaction and enhanced activity. The R4-position was close to the SER280 group, and the presence of substituents could form steric hindrance, which was a disadvantage factor for activity.
The R5-position of Ring C was located at the outer side of the active site cavity; thus, different substituents could be introduced to modify the structure. Different substituents may interact with different amino acid residues, but there was no obvious regularity between activity changes and various substituents.
The R6-position of Ring C was very close to the LYS337 residue, and the existence of substituents tended to form steric hindrance, reducing activity. For example, compared with Compound 13, the activity of Compound 17 with -COOC2H5 in the R6-position decreased significantly, and compared with Compound 23, the activity of Compound 20 with -NS2O4CH3 in the R6-position also decreased distinctly. Therefore, it was disadvantageous to add substituent groups in the R6-position in order to increase the activity of the compounds.
To summarize, the R1-, R4-, and R6-positions were close to amino acids HIS283, SER280, and LYS337 of the receptor, respectively, and the steric hindrance caused by the substituent group could significantly decrease the activity, which was an unfavorable site for structural modification of the compounds. The hydrophobic groups in the R2- or R3-position could increase hydrophobicity and enhance activity. However, the R5-position was near the outer side of the active site cavity, and so different substituent groups can be introduced in this position, modifying the structure to increase the activity or improve the physical and chemical properties of the inhibitors.
2.3. Design and Screening of New Inhibitors
On the basis of the analysis of the binding modes of ligands in the active site and structure-activity relationships, combined with the characteristics of pharmacophore [
12], 29,254 new compounds were designed for virtual screening using the reaction-based in situ enumeration method and scaffold hopping methods with DS software and experience-based manual design. To make the retrieved compounds more drug-like, the selected hit compounds were required to meet Lipinski’s rule of five. After that, a set of 9254 compounds were selected from the 29,254 compounds by Lipinski’s rule of five for the subsequent CDOCKER docking study. All compounds were docked into the active site of 6B8Y using CDOCKER docking. Novel inhibitors with a lower CDOCKER energy and a similar action mechanism were selected for the next study. Finally, five novel inhibitors (CQMU1901–1905) with potential activity were screened. Their structures are illustrated in
Figure 6, and relevant data are listed in
Table 4.
The novel inhibitors have the following characteristics: (1) Structural modification with purine as the mother nucleus, which is obviously different from the existing compound structure, can effectively break through patent protection; (2) like the existing molecules, the skeleton structures of the novel inhibitors have three aromatic rings which can form hydrophobic interaction with three regions of the receptor; (3) in addition to the hydrogen bonds between ligands and residues LYS232 and HIS283, the novel inhibitors can also form new hydrogen bonds with other amino acid residues in the active site, which is conducive to enhance the interaction between ligand and receptor.
Among them, CQMU1905 is a very interesting molecule for the following reasons: (1) It is composed of 5-fluorouracil (5-FU), 6-mercaptopurine (6-MP), and 5-azacytosine, and, as we all know, 5-FU, 6-MP, and 5-azacytidine are often used as anti-metabolic agents in cancer treatment; (2) as illustrated in
Figure 7B, compared with Ring A of Compound 1, the azacytosine ring of CQMU1905 can form N–H···O hydrogen bonds between -NH2 in azacytosine ring and residue LYS281, as well as hydrogen bonds between the N atom on the azacytosine ring and residue HIS283; compared with Ring B of Compound 1, two hydrogen bonds were formed between carbonyl group in 5-FU of CQMU1905 and residue ASP351 and TYR249. Aside from this, there are halogen bonds between the F atom and residue ALA230 and LEU278, and compared with Ring C, the 6-MP of CQMU1905 not only forms hydrogen bonds with residue LYS232, it also forms hydrogen bonds with residue ASP351; (3) the -NH
2 group of azacytosine is located at an excellent site to modify the molecular, chemical, and metabolic properties because the -NH
2 group can form acylamides easily with fatty acids—thus, the properties of molecules such as LogP can be improved, the deamination effect can be reduced, and the reaction time can be increased.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






















