mRNA and miRNA Expression Analysis Reveal the Regulation for Flower Spot Patterning in Phalaenopsis ‘Panda’


Abstract
:
1. Introduction
2. Results
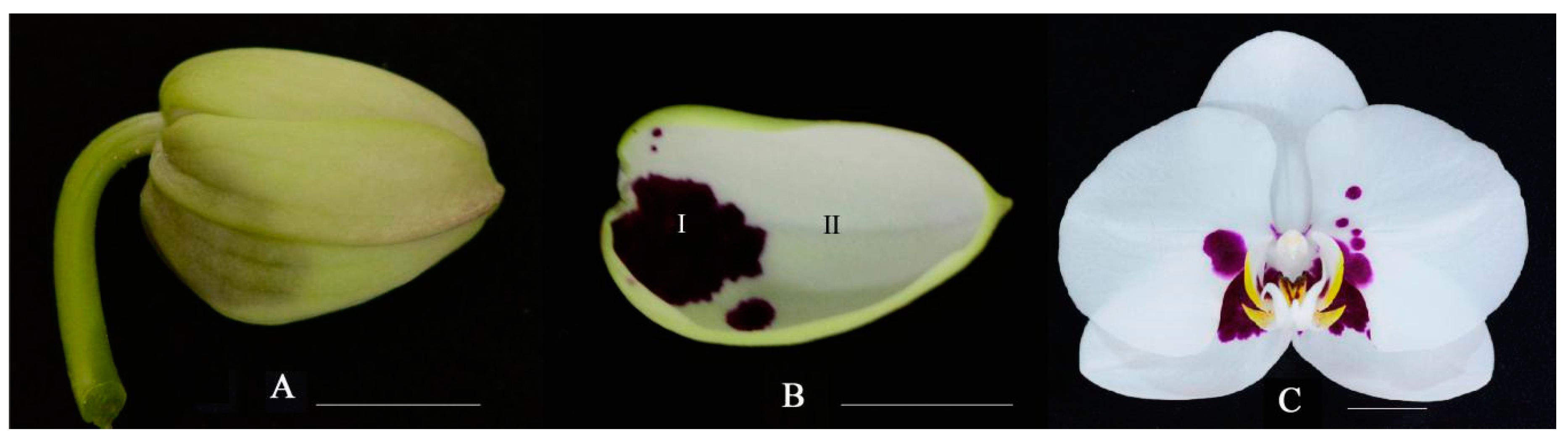
2.1. Anthocyanin Accumulation Patterns in Phalaenopsis ‘Panda’
2.2. Construction of cDNA Library and Gene Mapping to the Reference Genomes
2.3. Functional Annotation and Classification
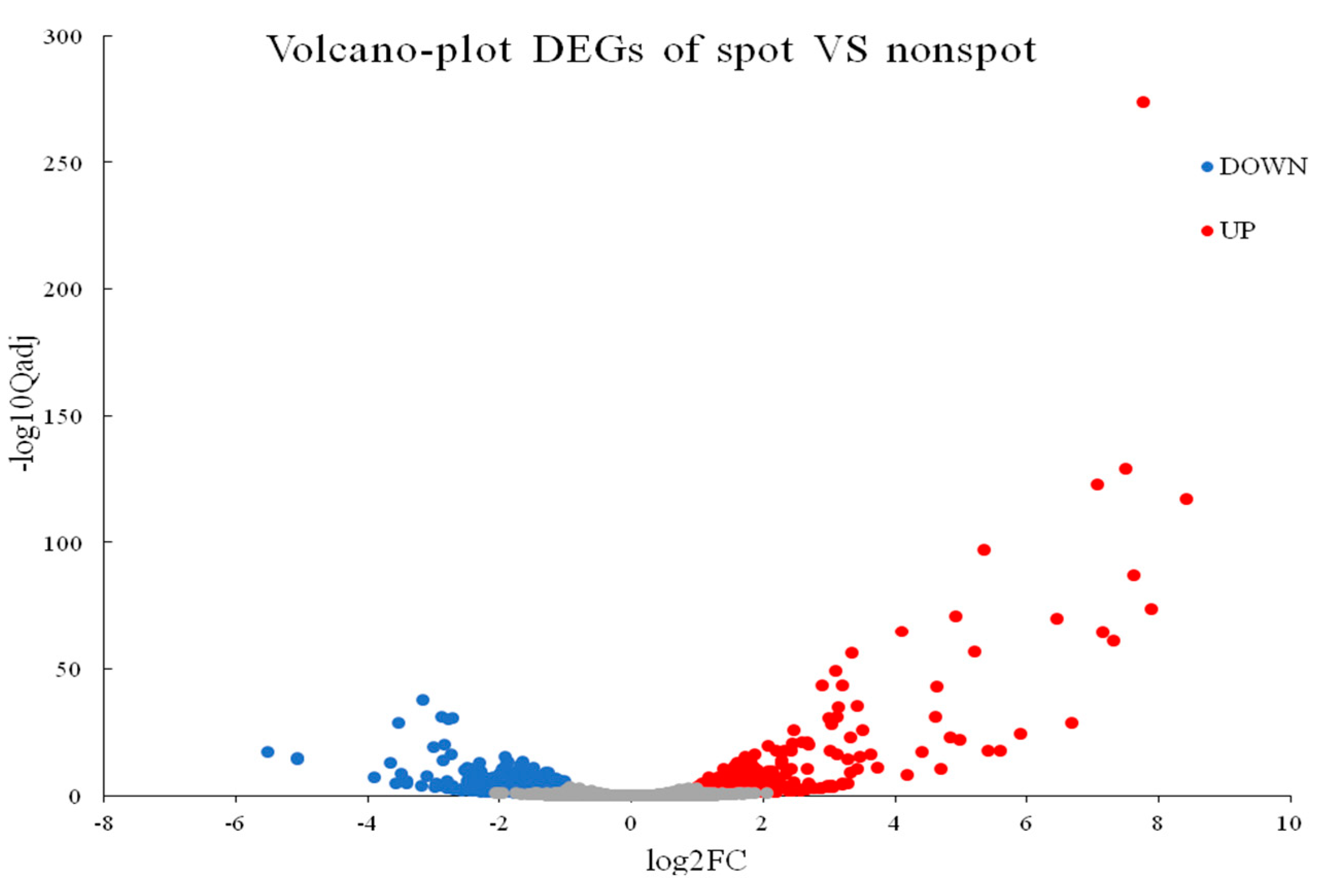
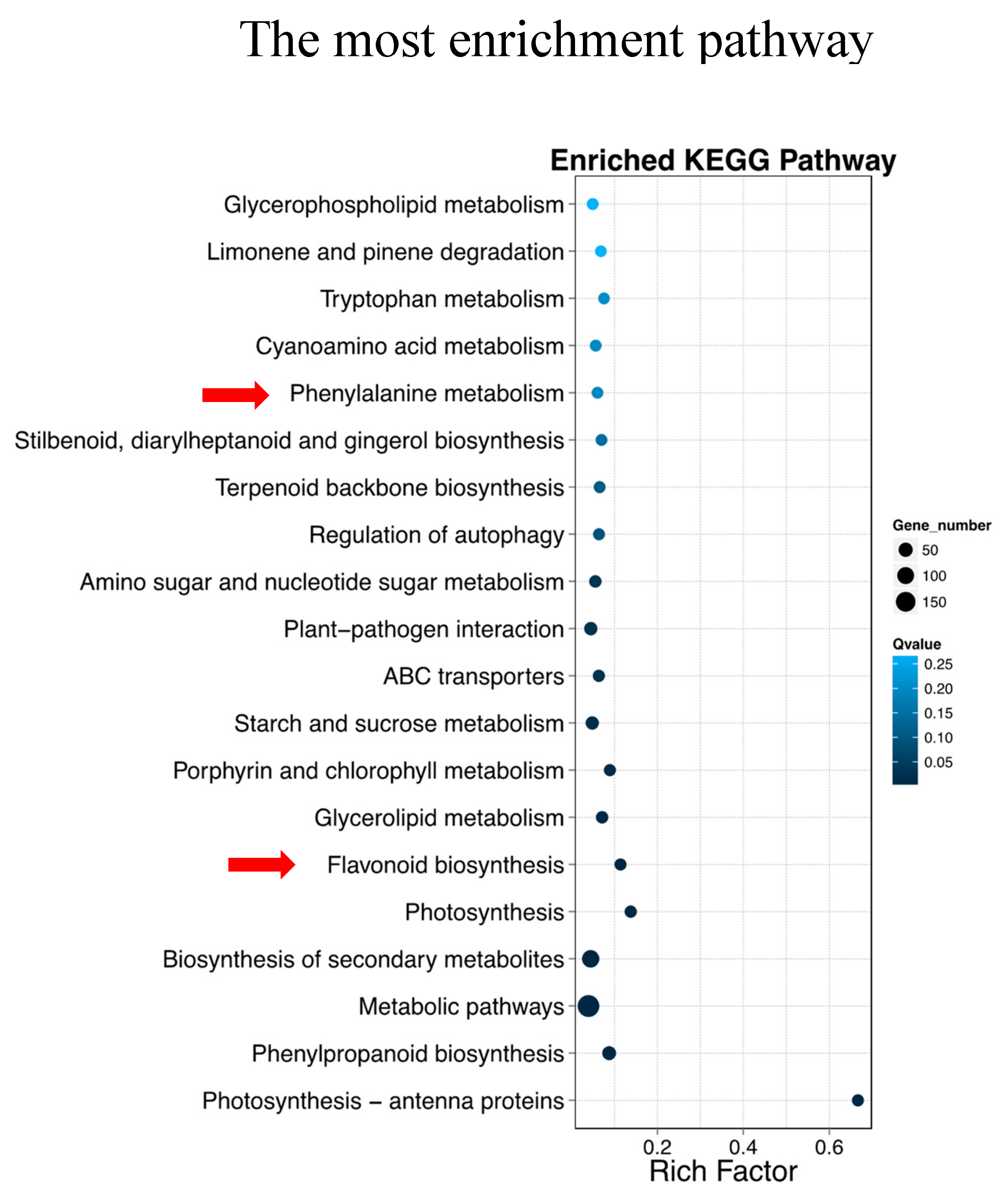
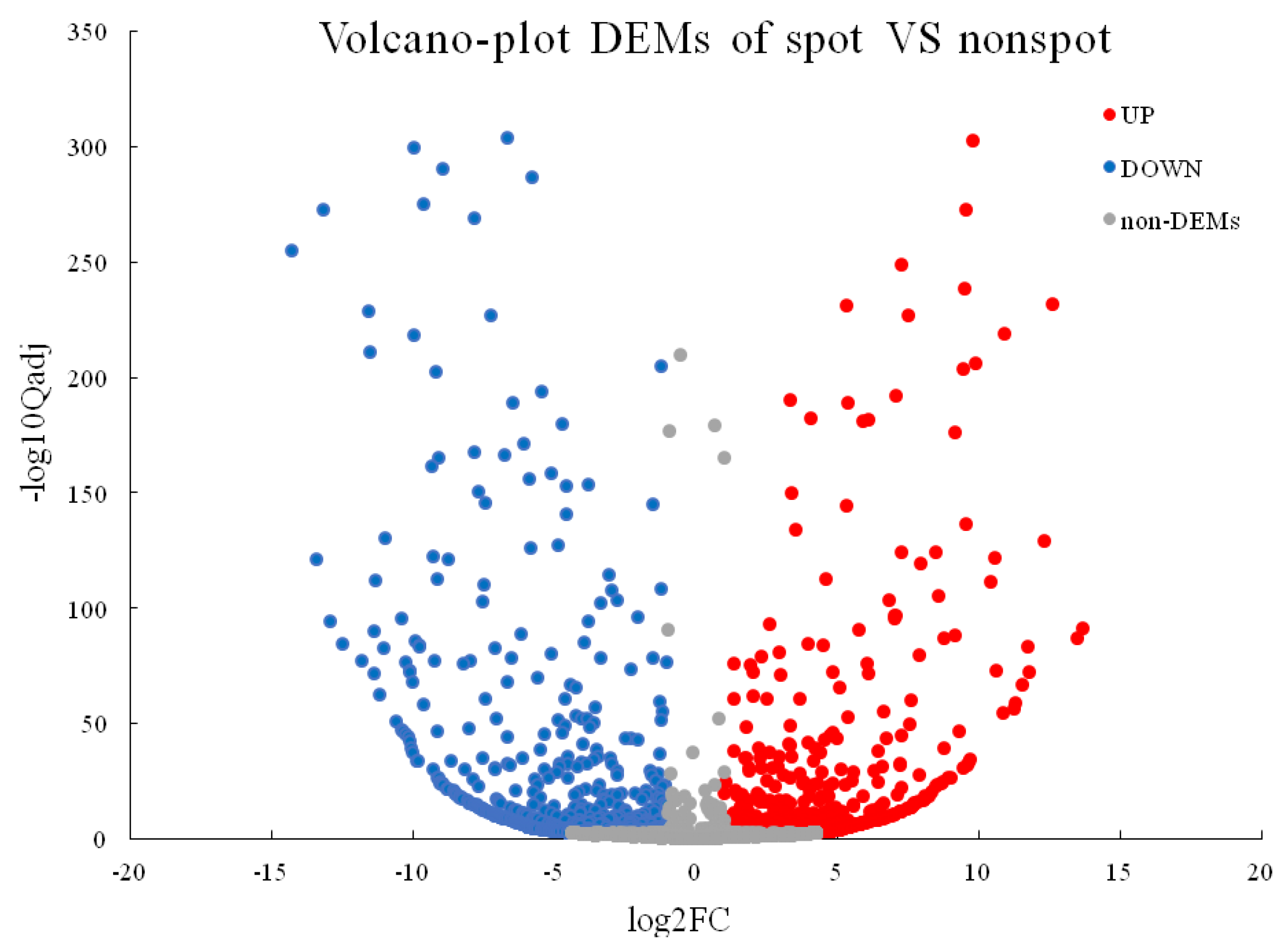
2.4. Identification of Differentially Expressed Genes and KEGG Enrichment Analysis of DEGs
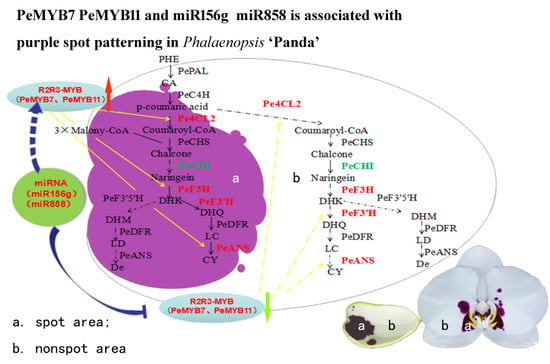
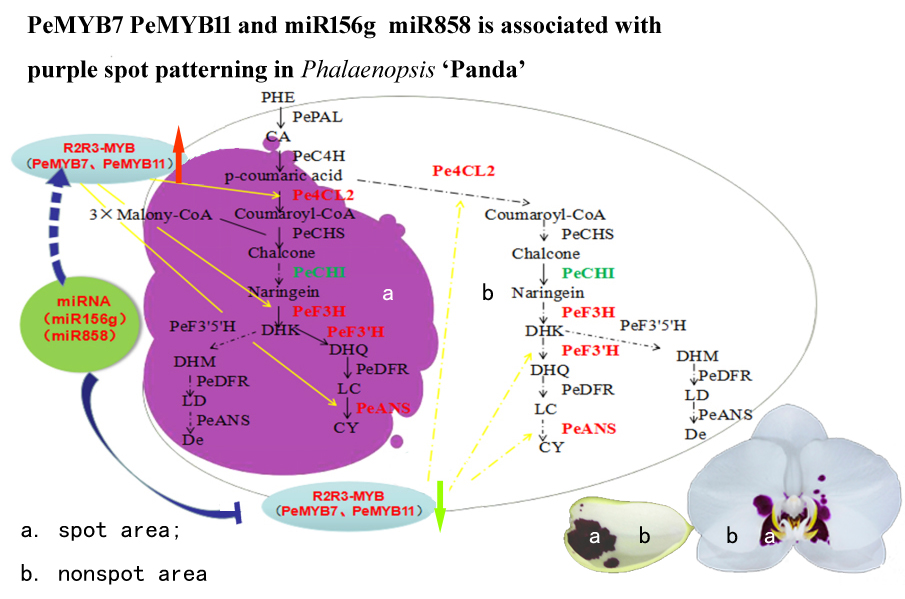
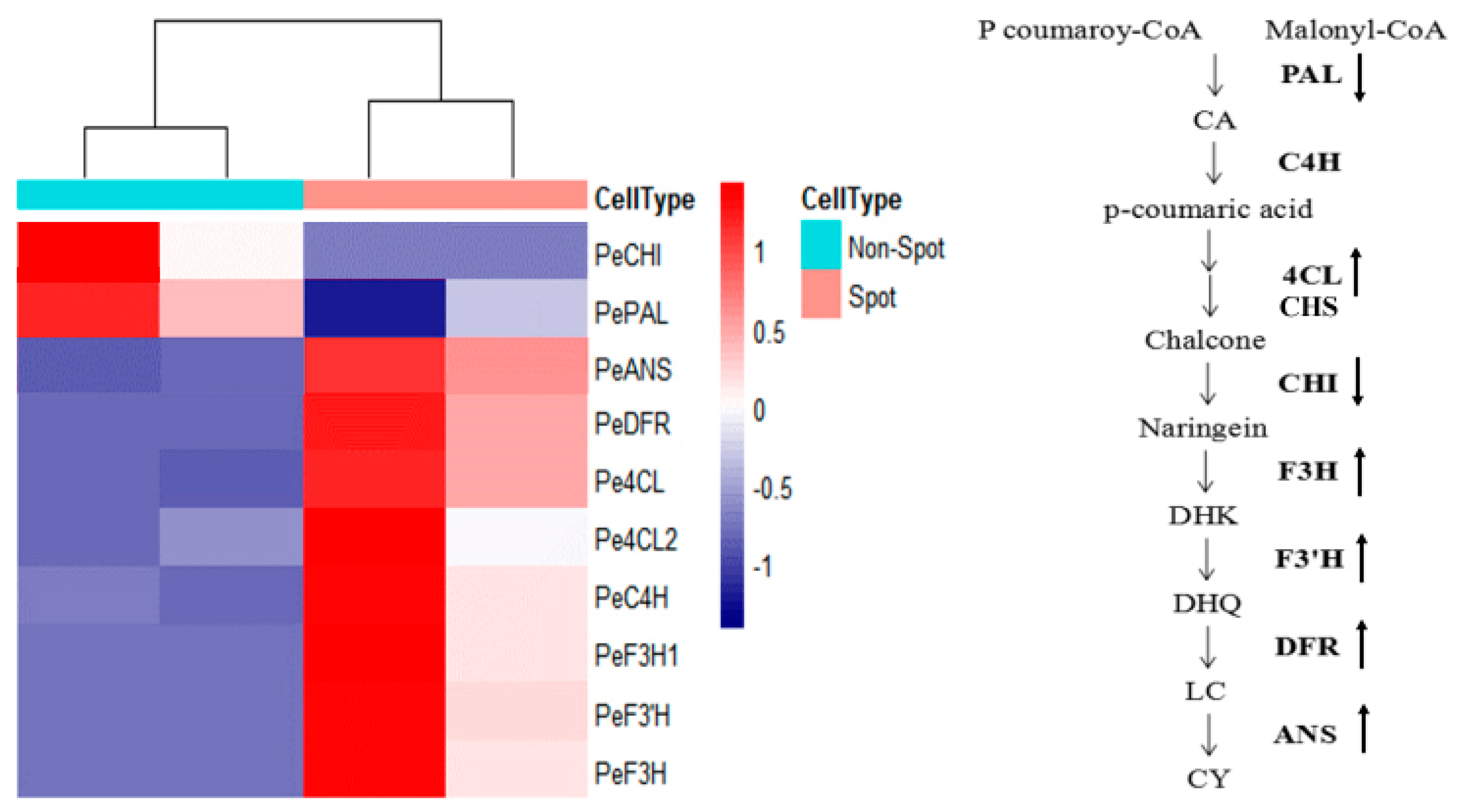
2.5. DEGs in Anthocyanin Biosynthesis and MBW Genes
2.6. Data Analysis of Small RNA Sequencing
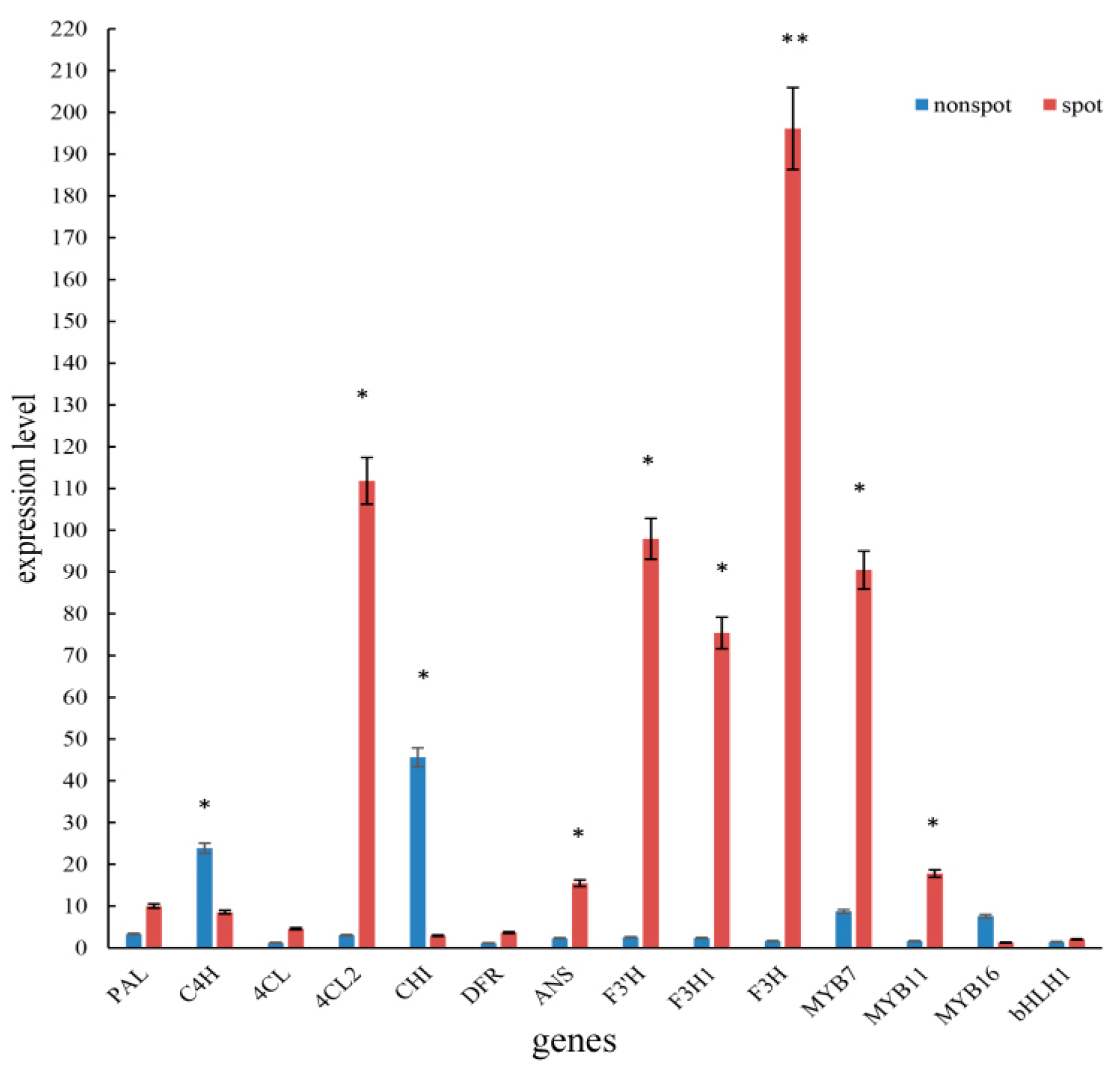
2.7. qPCR of Key Structural Genes, Regulate Genes and miRNA
3. Discussion
3.1. Low Expression of Anthocyanin Genes Causing the Lack of Pigments in Non-Spot Areas
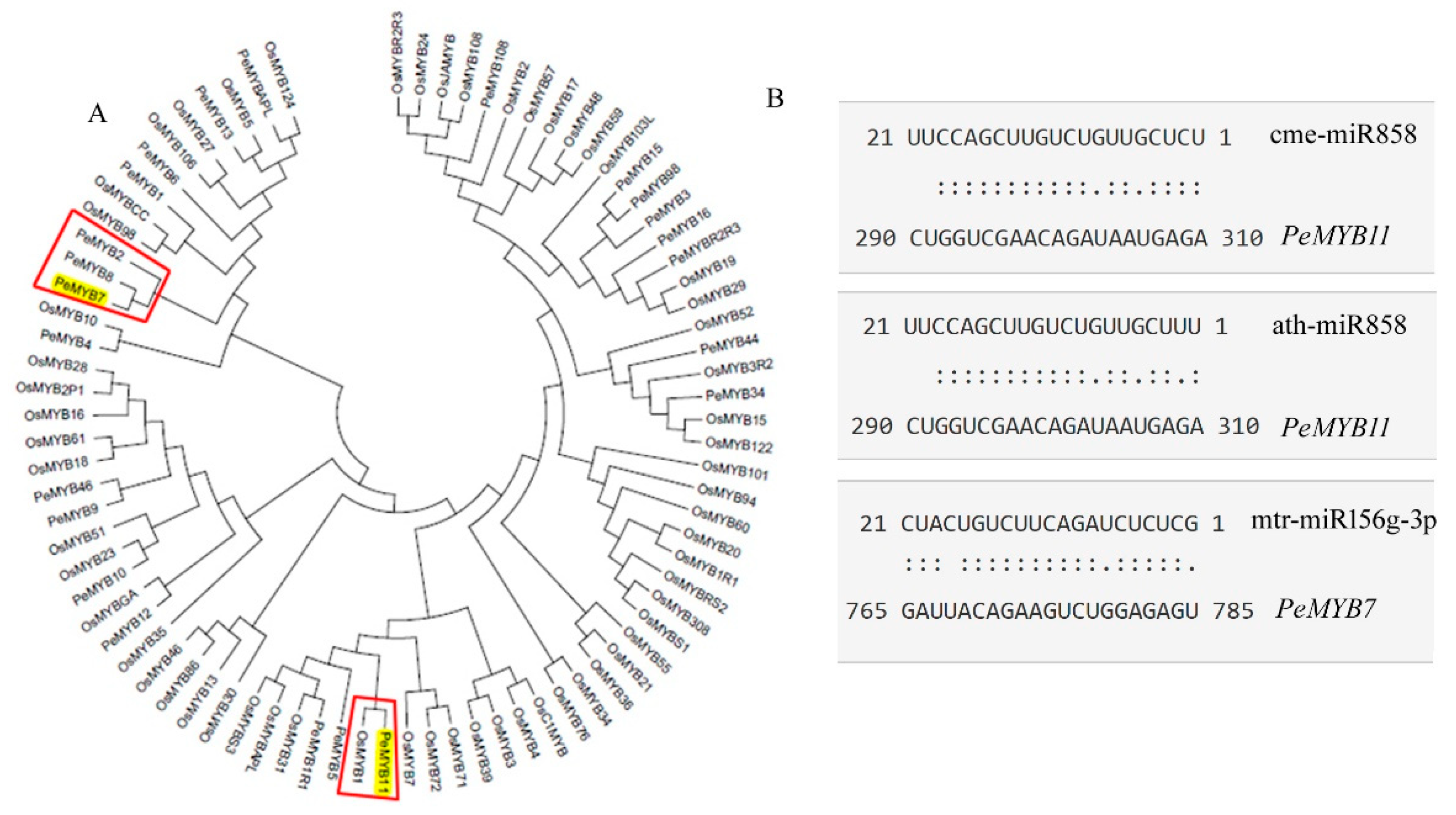
3.2. PeMYB7 and PeMYB11 Are Important Genes in Spot Formation
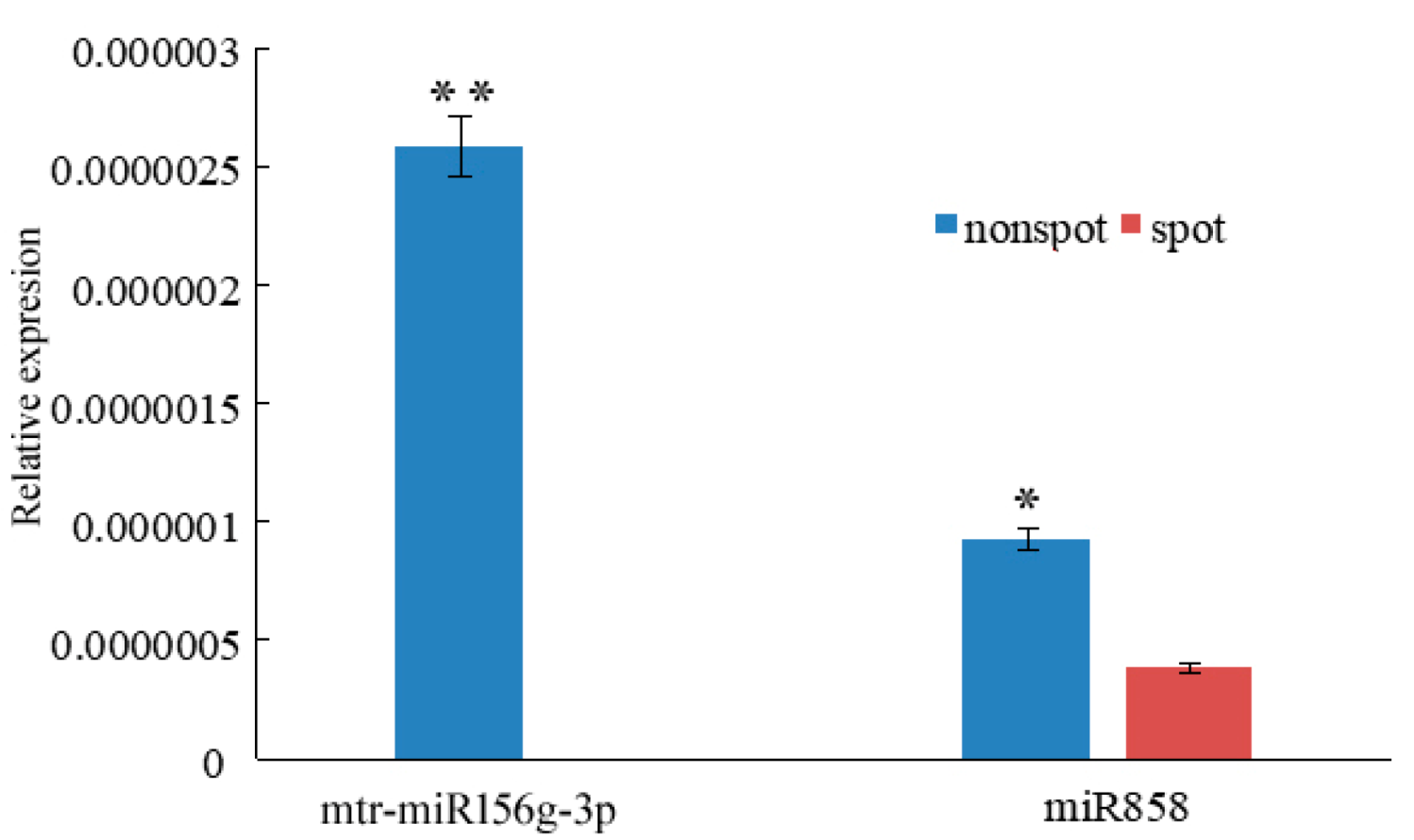
3.3. miR156g, miR858 Silence PeMYB7, and PeMYB11
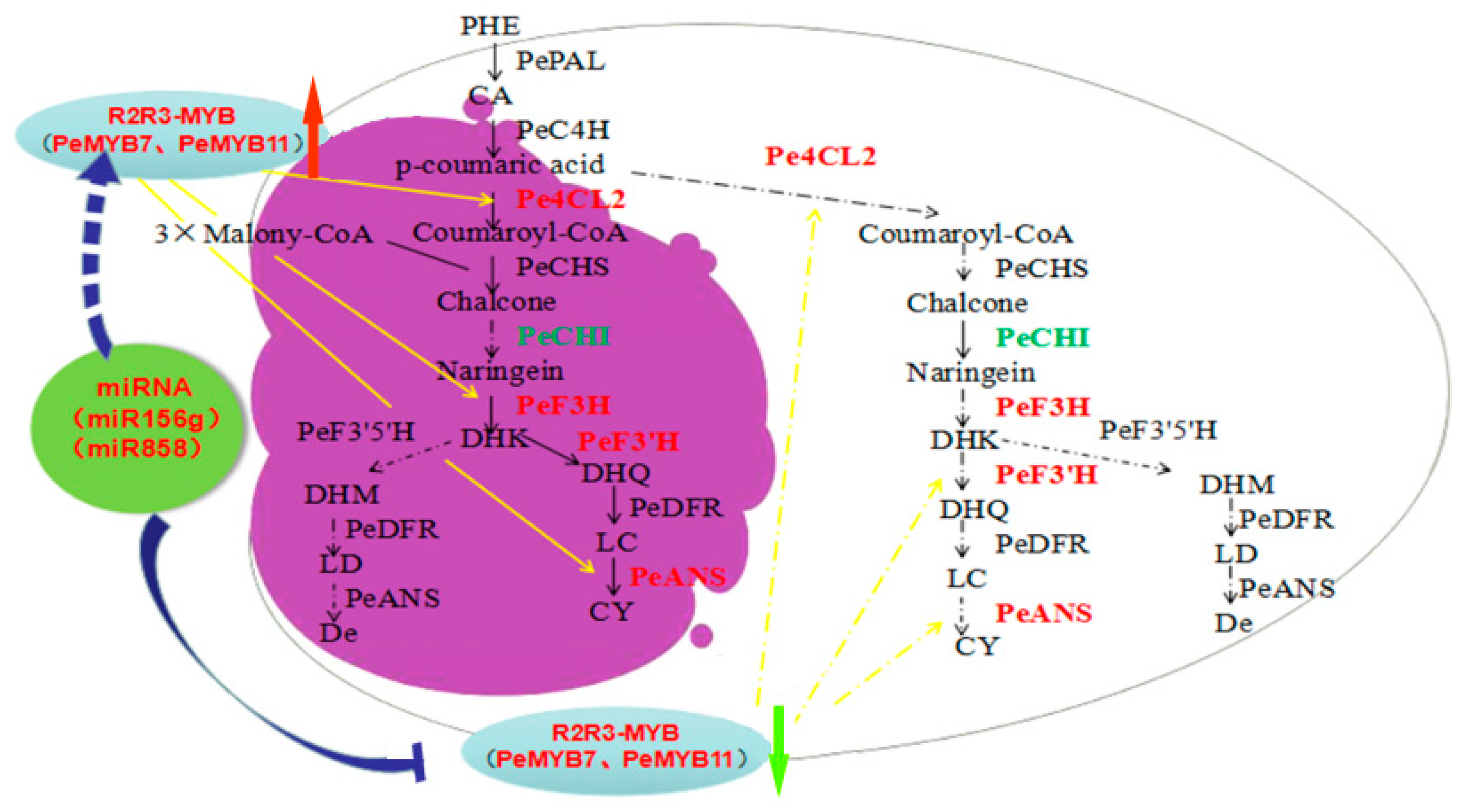
3.4. A Proposed Modelsummarizing of Spot Formation Pattern in Phalaenopsis ‘Panda’
4. Materials and Methods
4.1. Plant Materials
4.2. Observations of Sepal Anatomy and Determination of Total Anthocyanin Content
4.3. RNA Extraction, cDNA Library Construction, and mRNA Sequencing
4.4. mRNA Transcriptome Data Analysis
4.5. qRT-PCR Analyses of mRNA
4.6. Small RNA Library Construction and Sequencing
4.7. Data Analysis of Small RNA Sequencing
4.8. Verification of miRNA Expression Levels by qPCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Eckhart, V.M.; Rushing, N.S.; Hart, G.M.; Hansen, J.D. Frequency–dependent pollinator foraging in polymorphic Clarkia xantiana ssp. xantiana populations: Implications for flower colour evolution and pollinator interactions. Oikos 2010, 112, 412–421. [Google Scholar] [CrossRef]
- Miller, R.; Owens, S.J.; Rørslett, B. Plants and colour: Flowers and pollination. Opt. Laser Technol. 2011, 43, 282–294. [Google Scholar] [CrossRef]
- Moeller, D.A. Pollinator community structure and sources of spatial variation in plant--pollinator interactions in Clarkia xantiana ssp. xantiana. Oecologia 2005, 142, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Venail, J.; Mackay, S.; Bailey, P.C.; Schwinn, K.E.; Jameson, P.E.; Martin, C.R.; Davies, K.M. The molecular basis for venation patterning of pigmentation and its effect on pollinator attraction in flowers of Antirrhinum. New Phytol. 2011, 189, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Chiou, C.Y.; Wu, K.; Yeh, K.W. Characterization and promoter activity of chromoplast specific carotenoid associated gene (CHRC) from Oncidium Gower Ramsey. Biotechnol. Lett. 2008, 30, 1861–1866. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, J.; Sun, H.Y.; Shang, X. Flower color patterning in pansy (Viola x wittrockiana Gams.) is caused by the differential expression of three genes from the anthocyanin pathway in acyanic and cyanic flower areas. Plant Physiol. Biochem. 2014, 84, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Forkmann, G.; Dangelmayr, B. Genetic control of chalcone isomerase activity in flowers of Dianthus caryophyllus. Biochem. Genet. 1979, 18, 519–527. [Google Scholar] [CrossRef]
- Matin, C.; Prescott, A.; Mackay, S.; Bartlett, J.; Vrijlandt, E. Control of anthocyanin biosynthesis in flowers of Antirrhinum majus. Plant J. 1991, 1, 37–49. [Google Scholar]
- Holton, T.A.; Brugliera, F.; Tanaka, Y. Cloning and expression of flavonal synthase from Petunia hybrida. Plant J. 1993, 4, 1003–1010. [Google Scholar] [CrossRef]
- Kawabata, S.K.Y.; Kusuhara, Y.K.; Li, Y.H.; Sakiyama, R.Z. The regulation of anthocyanin Biosythesis in Eustoma grandiflorum under low light condition. J. Jpn. Soc. Hort. Sci. 1999, 68, 519–526. (In Japanese) [Google Scholar] [CrossRef]
- Bashandy, H.; Pietiainen, M.; Carvalho, E.; Lim, K.J.; Elomaa, P.; Martens, S.; Teeri, T.H. Anthocyanin biosynthesis in gerbera cultivar ‘Estelle’ and its acyanic sport ‘Ivory’. Planta 2015, 242, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.M.; Pooler, M.; Griesbach, R. Anthocyanin Regulatory/Structural Gene Expression in Phalaenopsis. J. Am. Soc. Hortic. Sci. 2009, 134, 88–96. [Google Scholar] [CrossRef]
- Katsumoto, Y.; Fukuchi-Mizutani, M.; Fukui, Y.; Brugliera, F.; Holton, T.A.; Karan, M.; Nakamura, N.; Yonekura-Sakakibara, K.; Togami, J.; Pigeaire, A. Engineering of the rose flavonoid biosynthetic pathway successfully generated blue-hued flowers accumulating delphinidin. Plant Cell Physiol. 2007, 48, 1589. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yonekura-Sakakibara, K.; Nakabayashi, R.; Higashi, Y.; Yamazaki, M.; Tohge, T.; Fernie, A.R. The flavonoid biosynthetic pathway in Arabidopsis: Structural and genetic diversity. Plant Physiol. Biochem. 2013, 72, 21–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Gao, L.; Wang, H.; Chen, X.; Wang, Y.; Yang, H.; Wei, C.; Wan, X.; Xia, T. The R2R3-MYB, bHLH, WD40, and related transcription factors in flavonoid biosynthesis. Funct. Integr. Genom. 2013, 13, 75–98. [Google Scholar] [CrossRef] [PubMed]
- Chiou, C.Y.; Yeh, K.W. Differential expression of MYB gene (OgMYB1) determines color patterning in floral tissue of Oncidium Gower Ramsey. Plant Mol. Biol. 2008, 66, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M. Oriental hybrid lily Sorbonne homologue of LhMYB12 regulates anthocyanin biosyntheses in flower tepals and tepal spots. Mol. Breed. 2011, 28, 381–389. [Google Scholar] [CrossRef]
- Whang, S.S.; Wan, S.U.; Song, I.J.; Lim, P.O.; Choi, K.; Park, K.W.; Kang, K.W.; Mi, S.C.; Koo, J.C. Molecular Analysis of Anthocyanin Biosynthetic Genes and Control of Flower Coloration by Flavonoid 3′,5′-Hydroxylase (F3′5′H) in Dendrobium moniliforme. J. Plant Biol. 2011, 54, 209–218. [Google Scholar] [CrossRef]
- Martins, T.R.; Berg, J.J.; Blinka, S.; Rausher, M.D.; Baum, D.A. Precise spatio-temporal regulation of the anthocyanin biosynthetic pathway leads to petal spot formation in Clarkia gracilis (Onagraceae). New Phytol. 2013, 197, 958–969. [Google Scholar] [CrossRef]
- Yamagishi, M.; Shimoyamada, Y.; Nakatsuka, T.; Masuda, K. Two R2R3-MYB genes, homologs of petunia AN2, regulate anthocyanin biosyntheses in flower tepals, tepal spots and leaves of Asiatic hybrid lily. Plant Cell Physiol. 2010, 51, 463–474. [Google Scholar] [CrossRef]
- Hsu, C.C.; Chen, Y.Y.; Tsai, W.C.; Chen, W.H.; Chen, H.H. Three R2R3-MYB transcription factors regulate distinct floral pigmentation patterning in Phalaenopsis spp. Plant Physiol. 2015, 168, 175–191. [Google Scholar] [CrossRef]
- Kaucsár, T.; Rácz, Z.; Hamar, P. Post-transcriptional gene-expression regulation by micro RNA (miRNA) network in renal disease. Adv. Drug Deliv. Rev. 2010, 62, 1390–1401. [Google Scholar] [CrossRef]
- Koseki, M.; Goto, K.; Masuta, C.; Kanazawa, A. The star-type color pattern in Petunia hybrida ‘red Star’ flowers is induced by sequence-specific degradation of chalcone synthase RNA. Plant Cell Physiol. 2005, 46, 1879–1883. [Google Scholar] [CrossRef]
- Guan, X.; Pang, M.; Nah, G.; Shi, X.; Ye, W.; Stelly, D.M.; Chen, Z.J. miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat. Commun. 2014, 5, 3050. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Liu, X.; Vanneste, K.; Proost, S.; Tsai, W.C.; Liu, K.W.; Chen, L.J. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 2015, 47, 65–72. [Google Scholar] [CrossRef]
- Xu, L.; Yang, P.; Feng, Y.; Xu, H.; Cao, Y.; Tang, Y.; Yuan, S.; Liu, X.; Ming, J. Spatiotemporal Transcriptome Analysis Provides Insights into Bicolor Tepal Development in Lilium “Tiny Padhye”. Front. Plant Sci. 2017, 8, 398. [Google Scholar] [CrossRef]
- Li, C.Q.; Shen, W.; Yan, P.; Li, X.Y.; Zhou, P. Bioinformatic Prediction of microRNA and Their Target Gene in Mimulus gutatus. Guizhou Agric. Sci. 2015, 1, 8–12. [Google Scholar]
- Zhang, Y.; Cheng, Y.; Ya, H.; Xu, S.; Han, J. Transcriptome sequencing of purple petal spot region in tree peony reveals differentially expressed anthocyanin structural genes. Front. Plant Sci. 2015, 6, 964. [Google Scholar] [CrossRef] [Green Version]
- Hsu, C.C.; Su, C.J.; Jeng, M.F.; Chen, W.H.; Chen, H.H. A HORT1 retrotransposon insertion in the PeMYB11 promoter causes harlequin/black flowers in Phalaenopsis orchids. Plant Physiol. 2019, 180, 1535–1548. [Google Scholar] [CrossRef]
- Hsu, C.C.; Chen, H.H. Flower Color and Pigmentation Patterns in Phalaenopsis Orchids. Orchid Biotechnology III; World Scientific Publishing: Singapore, 2017; pp. 393–420. [Google Scholar]
- Bonar, N.; Liney, M.; Zhang, R.; Austin, C.; Dessoly, J.; Davidson, D.; Stephens, J.; McDougall, G.; Taylor, M.; Bryan, G.J.; et al. Potato miR828 Is Associated With Purple Tuber Skin and Flesh Color. Front. Plant Sci. 2018, 9, 1742. [Google Scholar] [CrossRef] [Green Version]
- Gou, J.Y.; Felippes, F.F.; Liu, C.J.; Weigel, D.; Wang, J.W. Negative regulation of anthocyanin biosynthesis in Arabidopsis by a miR156-targeted SPL transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef]
- Chitwood, D.H.; Klein, L.L.; O’Hanlon, R.; Chacko, S.; Greg, M.; Kitchen, C.; Miller, A.; Londo, J.P. Latent developmental and evolutionary shapes embedded within the grapevine leaf. New Phytol. 2016, 210, 343–355. [Google Scholar] [CrossRef]
- Jia, X.; Shen, J.; Liu, H.; Li, F.; Ding, N.; Gao, C.; Pattanaik, S.; Patra, B.; Li, R.; Yuan, L. Small tandem target mimic-mediated blockage of microRNA858 induces anthocyanin accumulation in tomato. Planta 2015, 242, 283–293. [Google Scholar] [CrossRef]
- Ballester, A.R.; Molthoff, J.; Vos, R.D.; Hekkert, B.T.L.; Orzaez, D.; Fernándezmoreno, J.P.; Tripodi, P.; Grandillo, S.; Martin, C.; Heldens, J. Biochemical and molecular analysis of pink tomatoes: Deregulated expression of the gene encoding transcription factor SlMYB12 leads to pink tomato fruit color. Plant Physiol. 2010, 152, 71–84. [Google Scholar] [CrossRef]
- Giusti, M.M.; Wrolstad, R.E. Charaterization and Measurement of Anthocyanins by UV-Visible Spectroscopy. Food Anal. Chem. 2001. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Method 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−∆∆Ct Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef]
- Wu, H.J.; Ma, Y.K.; Chen, T.; Wang, M.; Wang, X.J. PsRobot: A web-based plant small RNA meta-analysis toolbox. Nucleic acids Res. 2012, 40, W22–W28. [Google Scholar] [CrossRef]
- Fahlgren, N.; Carrington, J.C. miRNA Target Prediction in Plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar]
- Varkonyi-Gasic, E.; Hellens, R.P. qRT-PCR of Small RNAs. Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; pp. 109–122. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Annotation | FPKM (non-spot) | FPKM (spot) | Log2FC | Padj | Up/Downregulation |
|---|---|---|---|---|---|---|
| PEQU_25924 | PeANS | 119.9303758 | 21,612.25078 | 7.493508409 | 4.71 × 10−130 | Up |
| PEQU_00400 | PeF3′H | 31.02321 | 6117.42 | 7.623432 | 5.88 × 10−88 | Up |
| PEQU_12025 | PeC4H | 2272.365 | 8325.431 | 1.87333 | 1.79 × 10−8 | Up |
| PEQU_34933 | PeDFR | 17.40537 | 5973.723 | 8.422954 | 1.13 × 10−117 | Up |
| PEQU_22606 | PeCHI | 2467.193 | 707.8831 | −1.80129 | 1.13 × 10−6 | Down |
| PEQU_07458 | Pe4CL | 11.87304 | 253.4331 | 4.415844 | 3.15 × 10−18 | Up |
| PEQU_38891 | PeF3H1 | 52.59874 | 5401.439 | 6.682172 | 2.59 × 10−29 | Up |
| PEQU_22432 | PeF3H | 21.49244 | 3076.48 | 7.161307 | 2.17 × 10−65 | Up |
| PEQU_01877 | PePAL | 14783.45 | 6368.878 | −1.21487 | 8.83 × 10−8 | Down |
| PEQU_00756 | Pe4CL2 | 383.56 | 1290.653 | 1.750578 | 0.000153 | Up |
| Gene ID | Annotation | FPKM (non-spot) | FPKM (spot) | Log2FC | Padj | Up/Downregulation |
|---|---|---|---|---|---|---|
| PEQU_03393 | PeMYB7 | 29.41538126 | 113.6604505 | 0.04965 | 0.00303 | UP |
| PEQU_10361 | PeMYB11 | 10.58835723 | 1693.628622 | 7.32149 | 8.71 × 10−62 | UP |
| PEQU_10362 | PeMYB11 | 4.49564678 | 216.34208328 | 5.58864183 | 2.35 × 10−18 | UP |
| PEQU_09064 | PeMYB16 | 1022.11354 | 508.0418102 | −1.00853 | 0.03605 | DOWN |
| PEQU_19747 | PebHLH1 | 149.2074009 | 801.9464953 | 2.42618 | 2.57 × 10−18 | UP |
| miRNA id | miRNA Expression in Non-spot | miRNA Expression in Spot | Up/Down Regulation of miRNA | p-Value | Target Gene | Target Unigene ID | Up/Downregulation of Target Unigene in Spot |
|---|---|---|---|---|---|---|---|
| Novel-m1700-5p | 12 | 0 | UP | 0.000818887 | PeMYB39 | PEQU_22029 | NA |
| mtr-miR156g-3p | 49 | 1 | UP | 2.66 × 10−12 | PeMYB7 | PEQU_03393 | UP |
| Novel-m0210-3p | 82 | 10 | UP | 1.69 × 10−13 | PeMYB7 | PEQU_03393 | UP |
| cme-miR858 | 52 | 0 | UP | 1.57 × 10−14 | PeMYB11 | PEQU_10361 | UP |
| PeMYB11 | PEQU_10362 | UP | |||||
| PeMYB8 | PEQU_10866 | NA | |||||
| PeMYB12 | PEQU_20333 | NA | |||||
| ath-miR858 | 336 | 116 | UP | 4.67 × 10−34 | PeMYB11 | PEQU_10361 | UP |
| PeMYB11 | PEQU_10362 | UP | |||||
| PeMYB12 | PEQU_20333 | NA | |||||
| ata-miR528-3p | 12 | 0 | UP | 0.000818887 | PebHLH086 | PEQU_08299 | NA |
| zma-miR528a-3p | 49 | 1 | UP | 2.66 × 10−12 | PebHLH086 | PEQU_08299 | NA |
| osa-miR162b | 82 | 10 | UP | 1.69 × 10−13 | PebHLH13 | PEQU_33912 | NA |
| Novel-m0112-3p | 0 | 52 | DOWN | 1.57 × 10−14 | PebHLH | PEQU_26133 | NA |
| gma-miR169v | 54 | 0 | UP | 2.83 × 10−13 | katanin p80 WD40 | PEQU_05516 | NA |
| Novel-m0290-5p | 24 | 0 | UP | 9.92 × 10−7 | katanin p80 WD40 | PEQU_05516 | NA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, A.; Cui, Z.; Li, T.; Pei, H.; Sheng, Y.; Li, X.; Zhao, Y.; Zhou, Y.; Huang, W.; Song, X.; et al. mRNA and miRNA Expression Analysis Reveal the Regulation for Flower Spot Patterning in Phalaenopsis ‘Panda’. Int. J. Mol. Sci. 2019, 20, 4250. https://doi.org/10.3390/ijms20174250
Zhao A, Cui Z, Li T, Pei H, Sheng Y, Li X, Zhao Y, Zhou Y, Huang W, Song X, et al. mRNA and miRNA Expression Analysis Reveal the Regulation for Flower Spot Patterning in Phalaenopsis ‘Panda’. International Journal of Molecular Sciences. 2019; 20(17):4250. https://doi.org/10.3390/ijms20174250
Chicago/Turabian StyleZhao, Anjin, Zheng Cui, Tingge Li, Huiqin Pei, Yuhui Sheng, Xueqing Li, Ying Zhao, Yang Zhou, Wenjun Huang, Xiqiang Song, and et al. 2019. "mRNA and miRNA Expression Analysis Reveal the Regulation for Flower Spot Patterning in Phalaenopsis ‘Panda’" International Journal of Molecular Sciences 20, no. 17: 4250. https://doi.org/10.3390/ijms20174250
APA StyleZhao, A., Cui, Z., Li, T., Pei, H., Sheng, Y., Li, X., Zhao, Y., Zhou, Y., Huang, W., Song, X., Peng, T., & Wang, J. (2019). mRNA and miRNA Expression Analysis Reveal the Regulation for Flower Spot Patterning in Phalaenopsis ‘Panda’. International Journal of Molecular Sciences, 20(17), 4250. https://doi.org/10.3390/ijms20174250