Transcriptome Analysis Reveals Key Seed-Development Genes in Common Buckwheat (Fagopyrum esculentum)


Abstract
:1. Introduction
2. Results
2.1. Overview of Sequencing Data Analysis
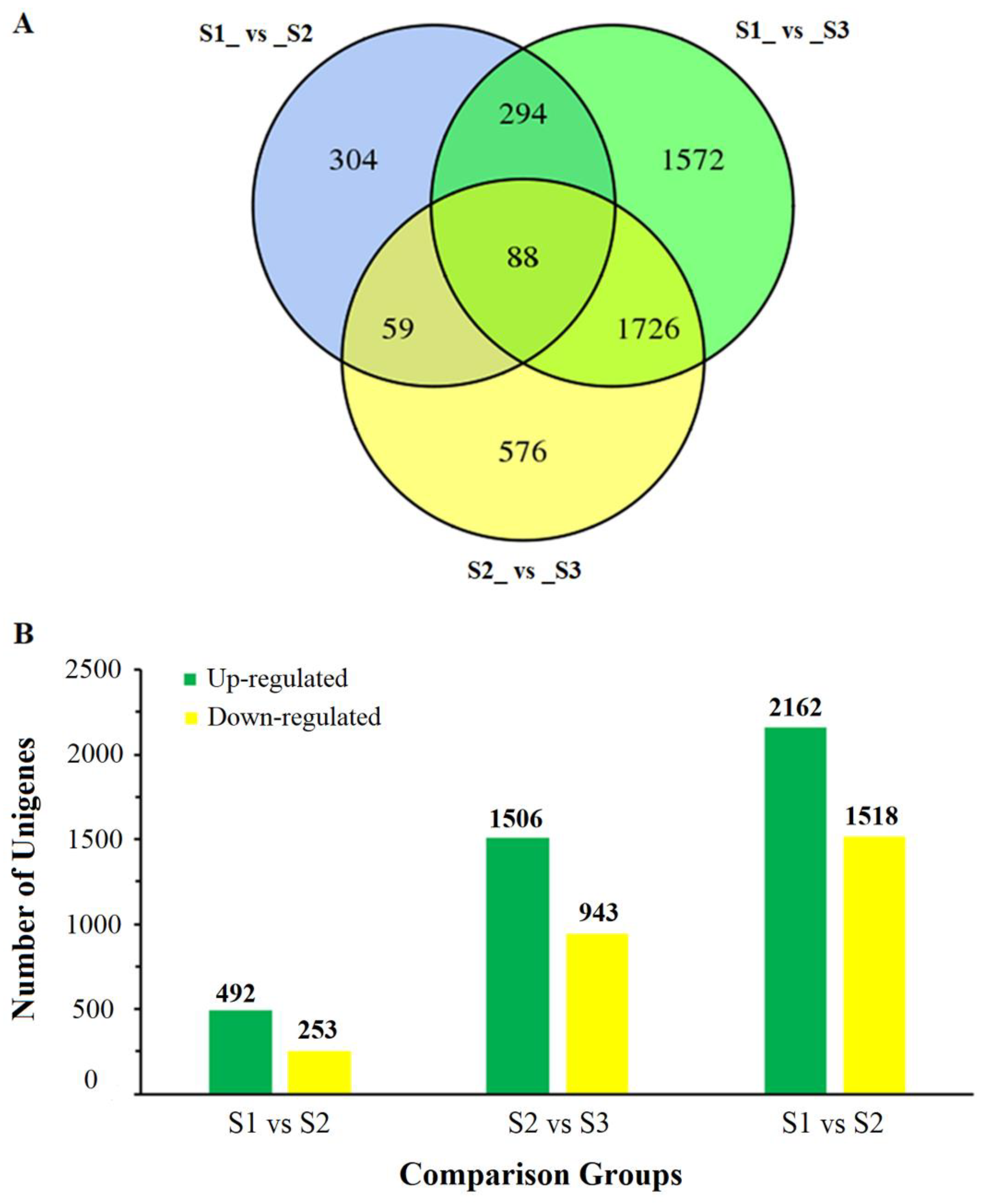
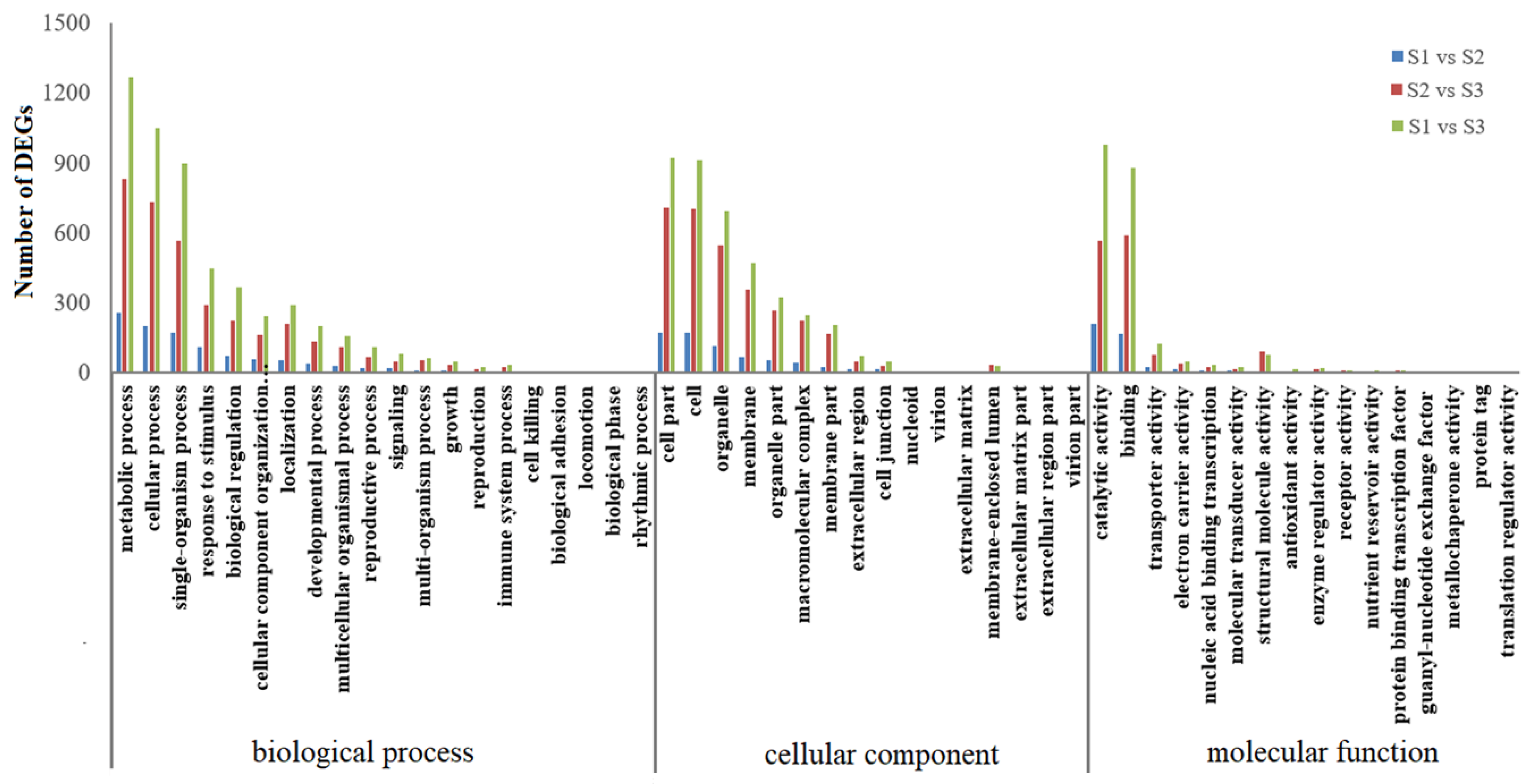
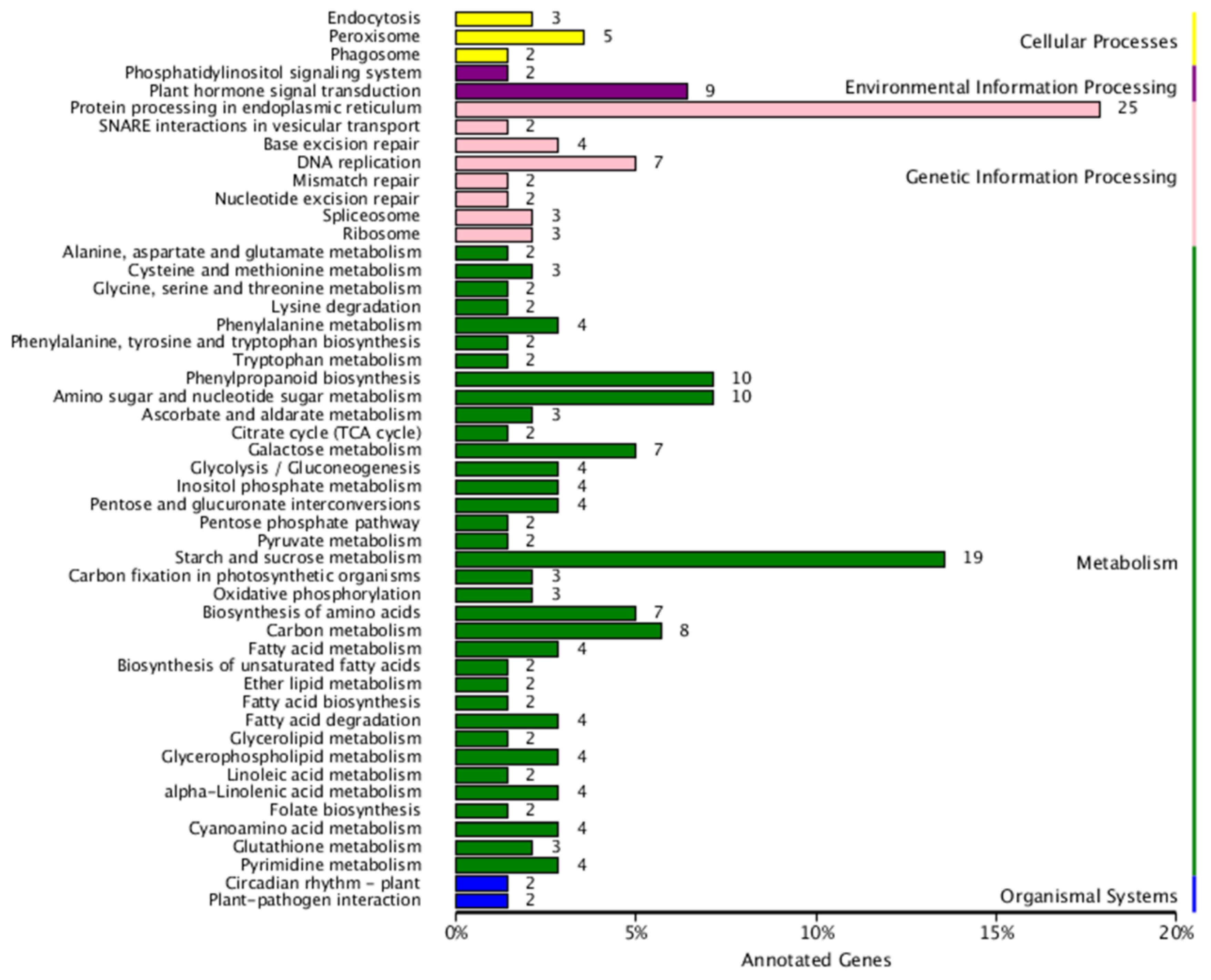
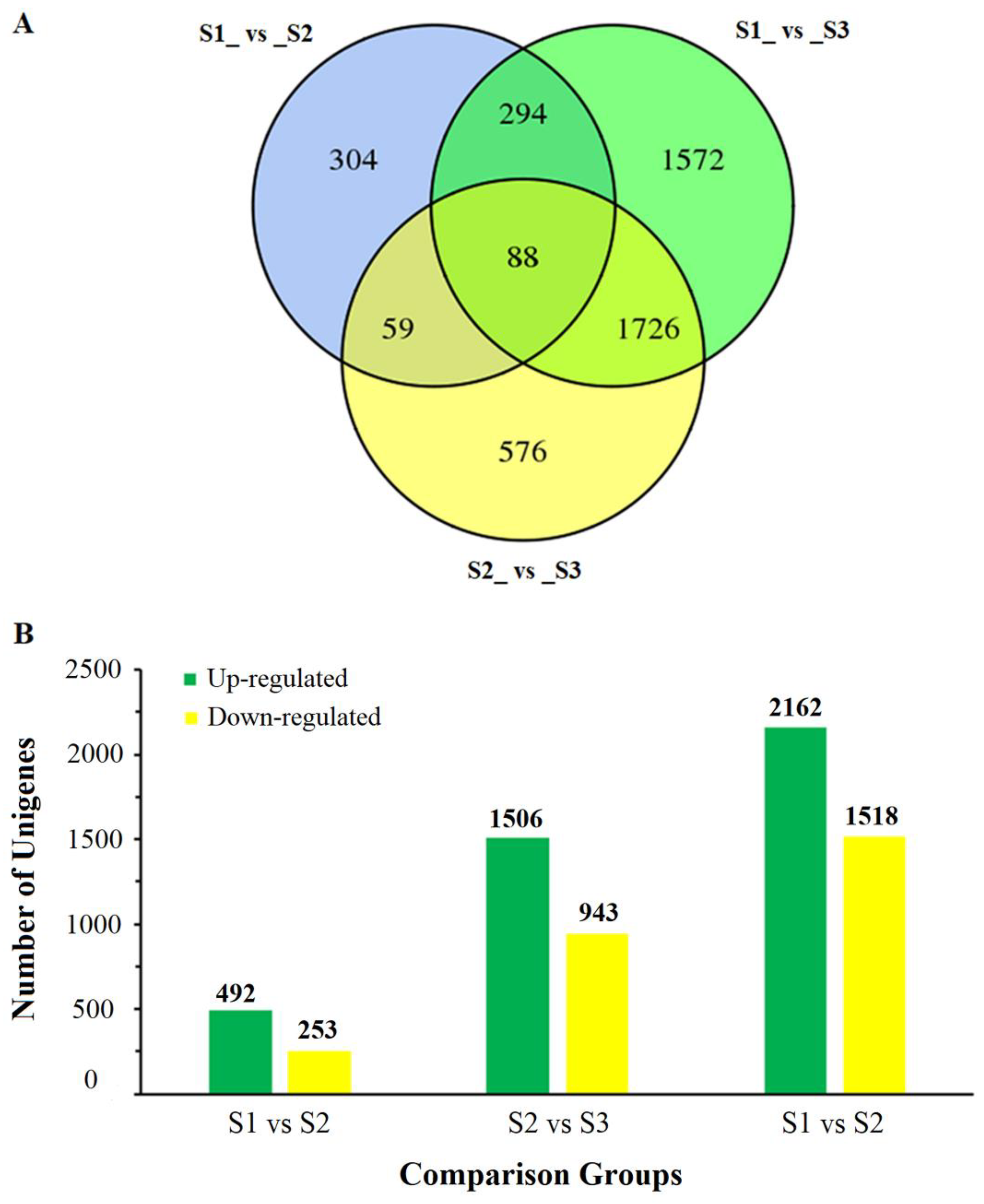
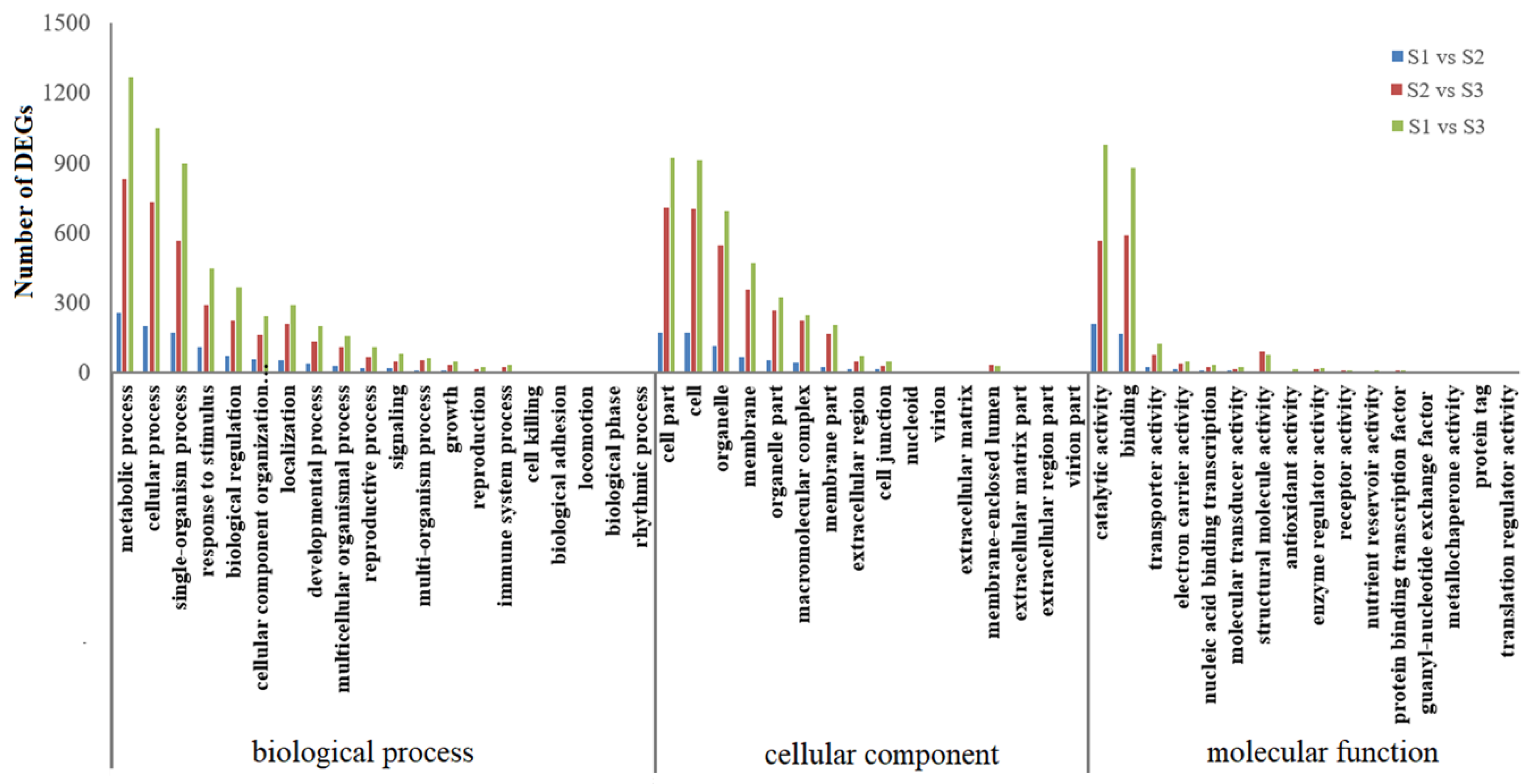
2.2. Identification and Annotation of Differentially Expressed Genes (DEGs)
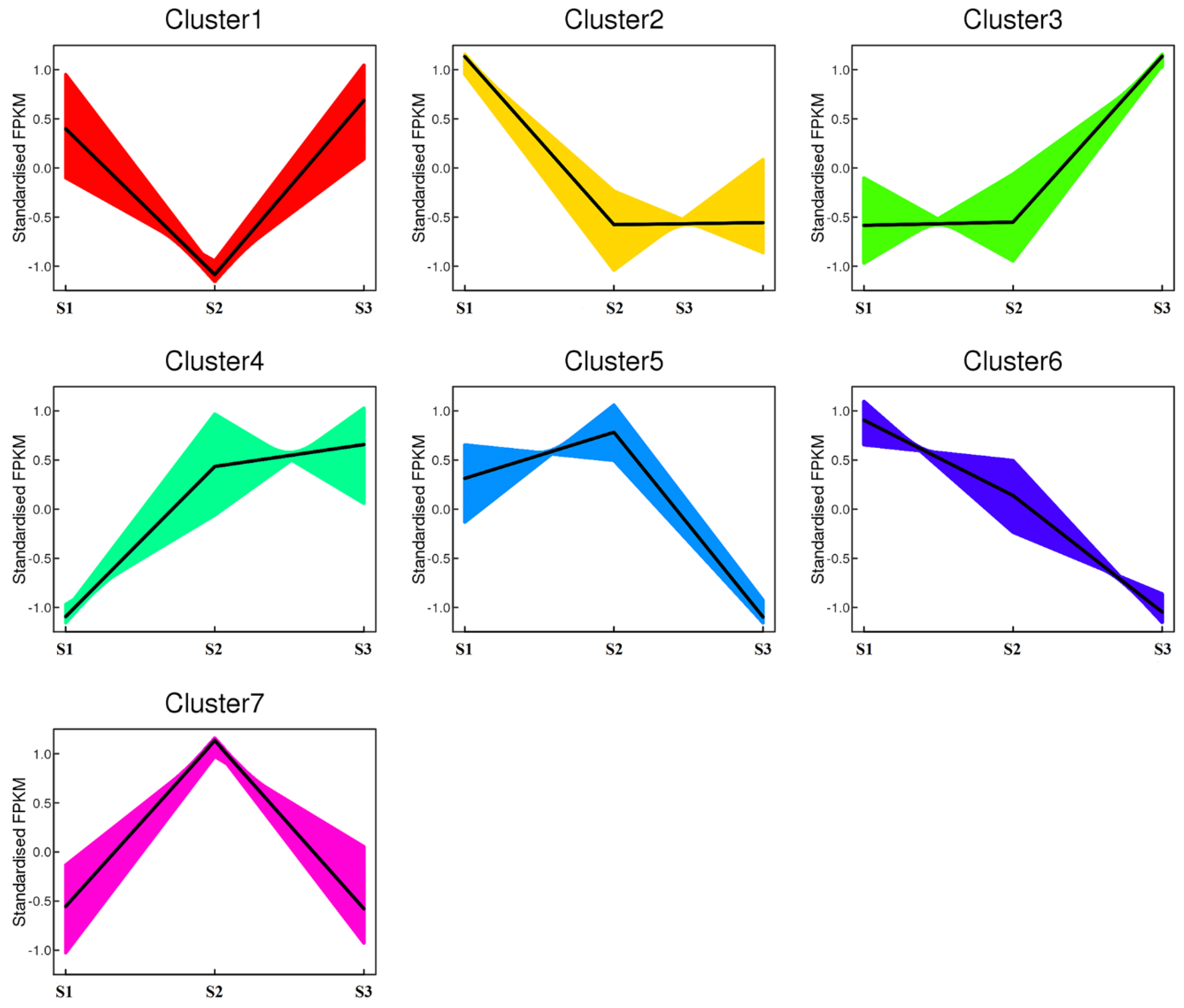
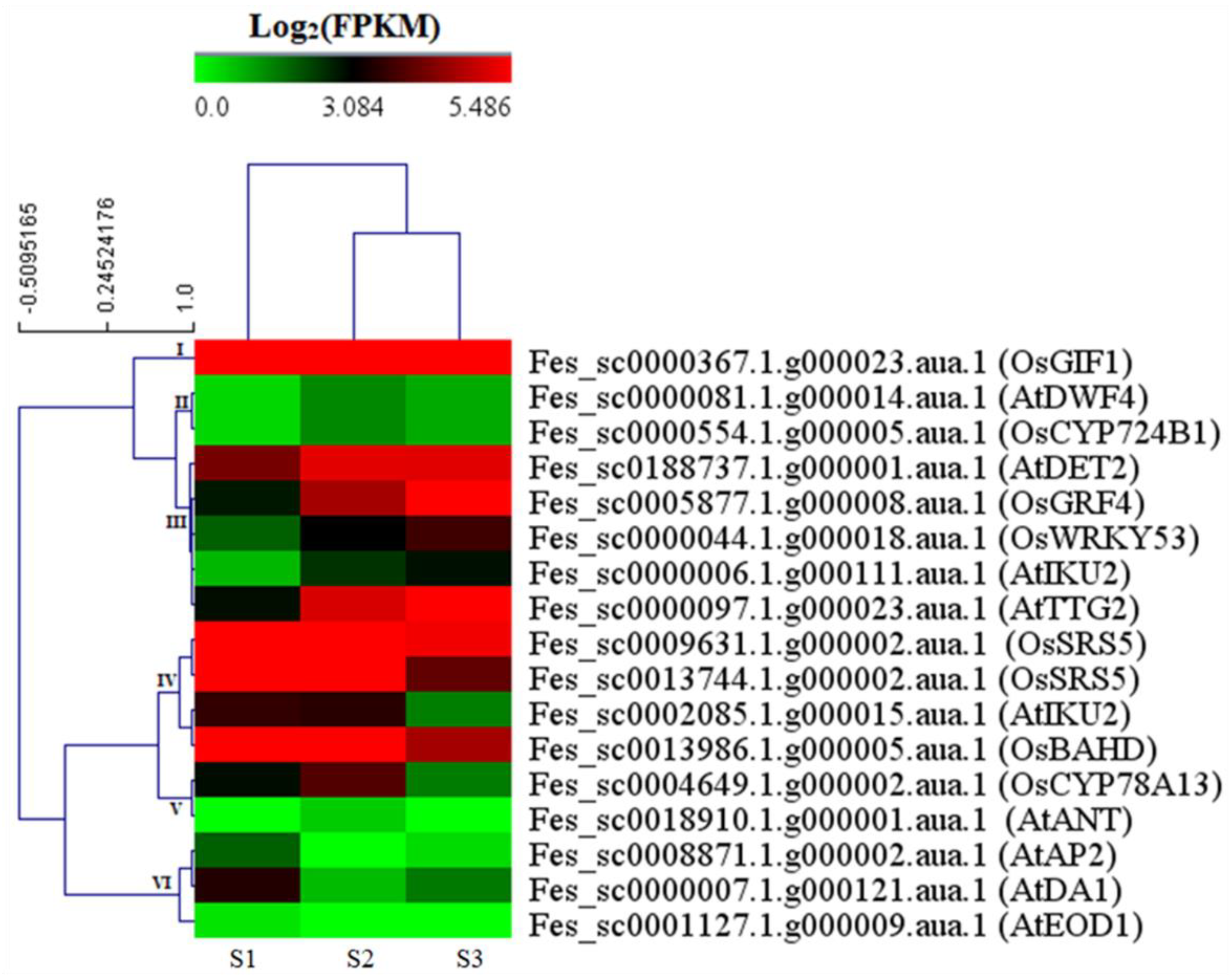
2.3. Key Genes Involved in the Seed Development of Common Buckwheat
2.3.1. DEGs Involved in Ca2+ Signal Transduction Pathways
2.3.2. DEGs Involved in Hormone Signal Transduction Pathways
2.3.3. DEGs Involved in TFs
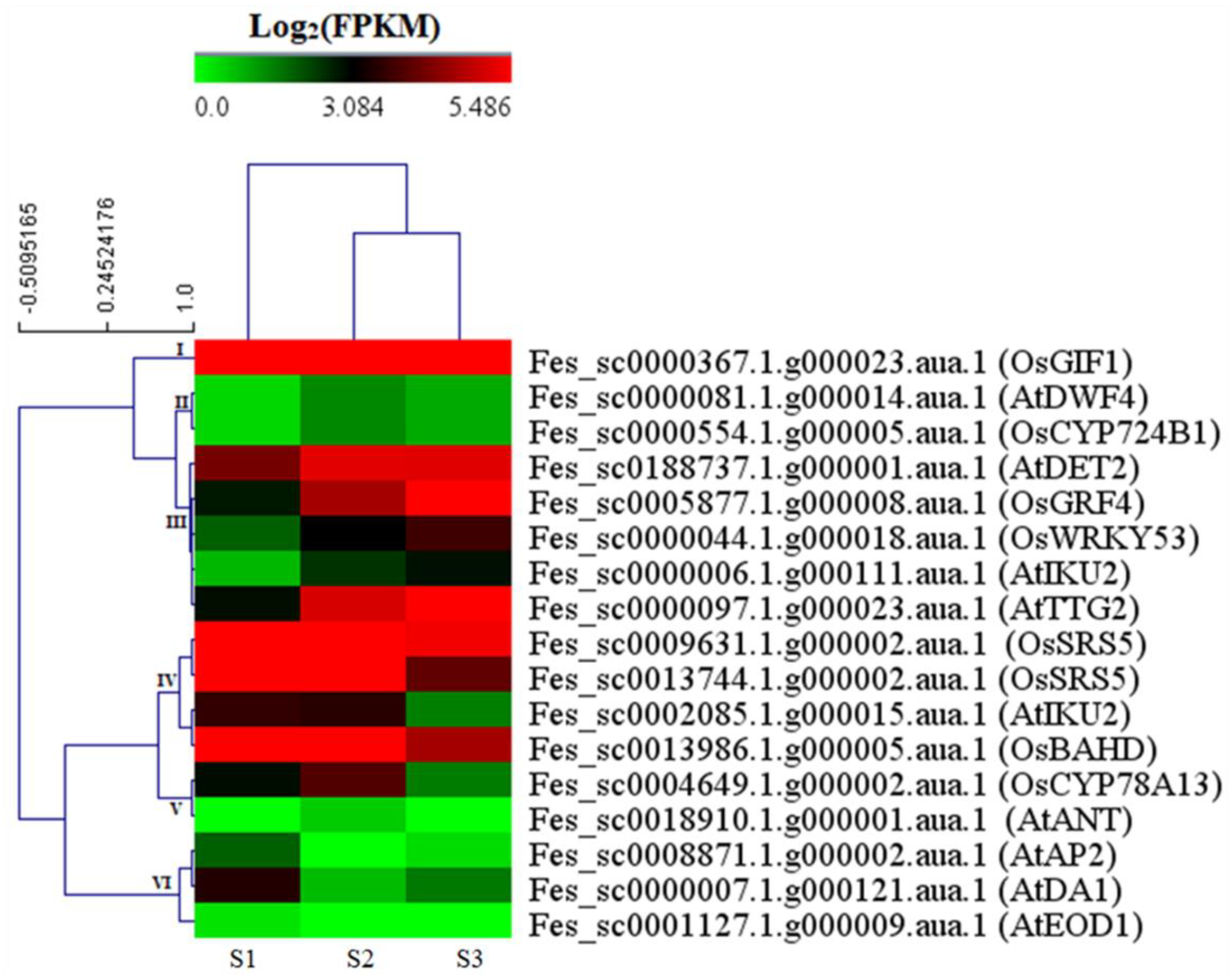
2.3.4. DEGs Involved in Seed Size
2.3.5. DEGs Involved in Starch Biosynthesis
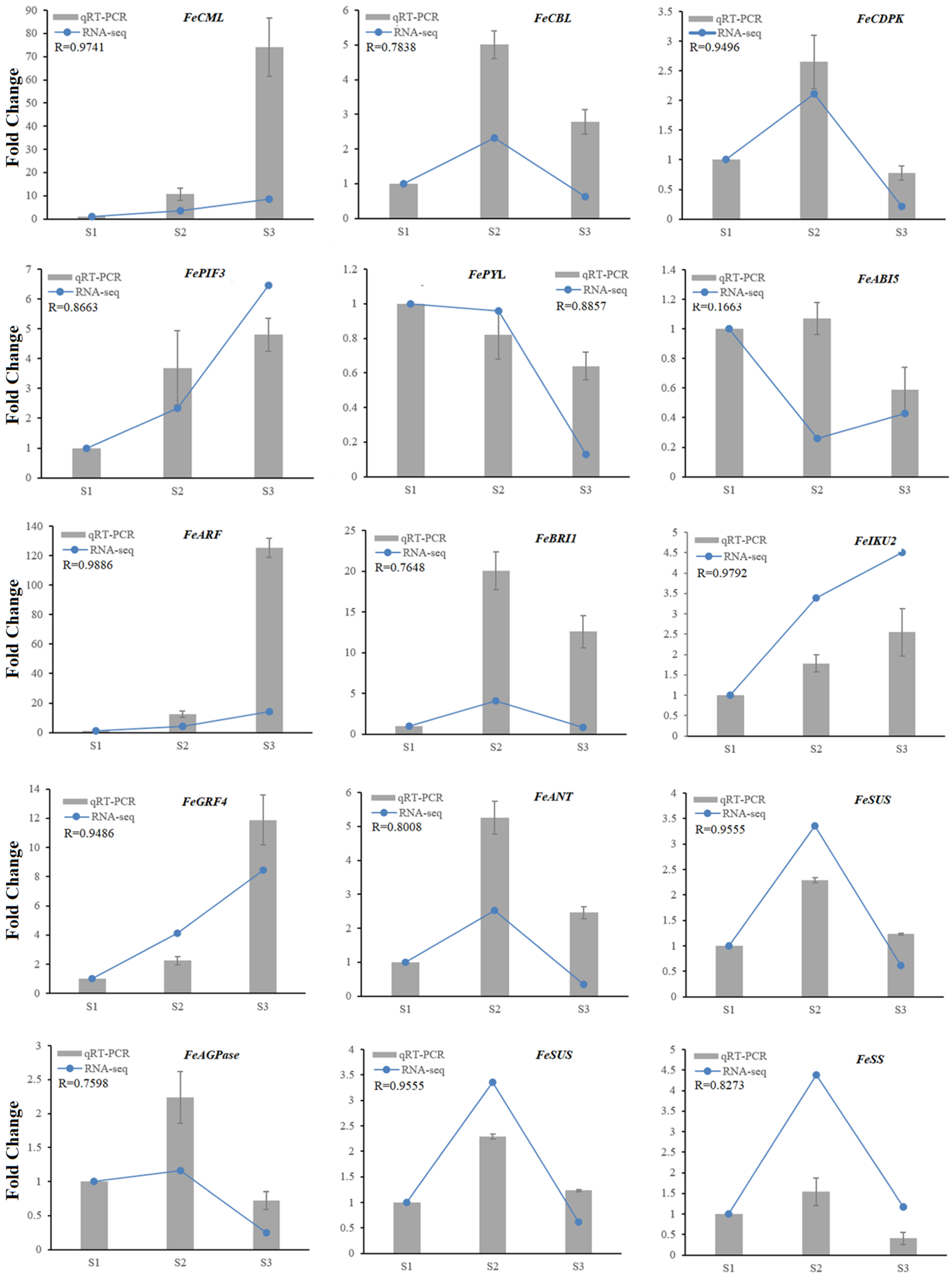
2.4. qRT-PCR Validation of RNA-Seq Results
3. Discussion
4. Materials and Methods
4.1. Plant Material and Sample Collection
4.2. RNA Extraction, Library Construction, and Sequencing
4.3. Analysis of RNA-Seq Data
4.4. Identification of DEGs
4.5. Validation of DEGs by qRT-PCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| ABF | ABA responsive element binding factor |
| AGPase | ADP glucose pyrophosphorylase |
| AHP | Histidine-containing phosphotransfer protein |
| BR | Brassinolides |
| BRI1 | Brassinosteroid insensitive 1 |
| CBLs | Calmodulin binding-like proteins |
| CCXs | Cation/Ca2+ exchangers |
| CDPKs | Ca2+-dependent protein kinases |
| CHX | Ca2+-transporting ATPase and Cation/H+ antiporter |
| IAA | Auxin-responsive protein |
| CK | Cytokinins |
| CMLs | Calmodulin-like proteins |
| CTR1 | Serine/threonine-protein kinase |
| DEB | Debranching enzyme |
| DEGs | Differentially expressed genes |
| ET | Ethylene |
| ETR | Ethylene Receptor |
| EIN3 | Ethylene-Insensitive Protein 3 |
| EIN4 | Ethylene-Insensitive Protein 4 |
| GA | Gibberellins |
| GBSS | Granule bound starch synthase |
| GO | Gene ontology |
| JA | Jasmonic acid |
| PIF3 | Phytochrome-interacting factor 3 |
| PP2C | Protein phosphatase 2C |
| PYR/PYL | Abscisic acid receptor |
| qRT-PCR | Quantitative real-time PCR |
| SA | Salicylic acid |
| SBE | Starch-branching enzyme |
| SnRK2 | Serine/threonine protein kinase |
| SS | Starch synthase |
| SUS | Sucrose synthase |
| TFs | Transcription factors |
| UGPase | UDP glucose pyrophosphorylase |
| QTLs | Quantitative trait locus |
References
- Kong, L.; Guo, H.; Sun, M. Signal transduction during wheat grain development. Planta 2015, 241, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Savadi, S. Molecular regulation of seed development and strategies for engineering seed size in crop plants. Plant Growth Regul. 2018, 84, 401–422. [Google Scholar] [CrossRef]
- Locascio, A.; Roig-Villanova, I.; Bernardi, J.; Varotto, S. Current perspectives on the hormonal control of seed development in Arabidopsis and maize: a focus on auxin. Front. Plant Sci. 2014, 5, 412. [Google Scholar] [CrossRef] [PubMed]
- Bian, J.; Deng, P.; Zhan, H.; Wu, X.; Nishantha, M.D.L.C.; Yan, Z.; Du, X.; Nie, X.; Song, W. Transcriptional dynamics of grain development in barley (Hordeum vulgare L.). Int. J. Mol. Sci. 2019, 20, 962. [Google Scholar] [CrossRef] [PubMed]
- Ruuska, S.A.; Girke, T.; Benning, C.; Ohlrogge, J.B. Contrapuntal networks of gene expression during Arabidopsis seed filling. Plant Cell 2002, 4, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Tzafrir, I.; Pena-Muralla, R.; Dickerman, A.; Berg, M.; Rogers, R.; Hutchens, S.; Sweeney, T.C.; McElver, J.; Aux, G.; Patton, D.; et al. Identification of genes required for embryo development in Arabidopsis. Plant Physiol. 2004, 135, 1206–1220. [Google Scholar] [CrossRef] [PubMed]
- Devic, M. The importance of being essential: EMBRYO-DEFECTIVE genes in Arabidopsis. C.R. Biol. 2008, 331, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, W.; Chen, Y.; Tang, W.; Yang, J.; Ye, R.; Liu, L.; Lin, Y.; Xu, C.; Xiao, J.; et al. A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 2010, 61, 752–766. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xu, R.; Li, Y. Molecular networks of seed size control plants. Annu. Rev. Plant Biol. 2019, 70, 435–463. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zeng, B.; Zhang, M.; Xie, S.; Wang, G.; Hauck, A.; Lai, J. Dynamic transcriptome landscape of maize embryo and endosperm development. Plant Physiol. 2014, 166, 252–264. [Google Scholar] [CrossRef]
- Li, G.; Wang, D.; Yang, R.; Logan, K.; Chen, H.; Zhang, S.; Skaggs, M.I.; Lloyd, A.; Burnett, W.J.; Laurie, J.D.; et al. Temporal patterns of gene expression in developing maize endosperm identified through transcriptome sequencing. Proc. Natl. Acad. Sci. USA 2014, 111, 7582–7587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantu, D.; Pearce, S.P.; Distelfeld, A.; Christiansen, M.W.; Uauy, C.; Akhunov, E.; Fahima, T.; Dubcovsky, J. Effect of the down-regulation of the high grain protein content (GPC) genes on the wheat transcriptome during monocarpic senescence. BMC Genomics 2011, 12, 492. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Gao, X.; Li, X.Y.; Chen, Q.J.; Dong, J.; Zhao, W.C. Evaluation of assembly strategies using RNA-seq data associated with grain development of wheat (Triticum aestivum L.). PLoS ONE 2013, 8, e83530. [Google Scholar] [CrossRef]
- Jones, S.I.; Vodkin, L.O. Using RNA-Seq to profile soybean seed development from fertilization to maturity. PLoS ONE 2013, 8, e59270. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, Q.T.; Xiong, Q.; Li, W.; Bi, Y.D.; Lai, Y.C.; Liu, X.L.; Man, W.Q.; Zhang, W.K.; Ma, B.; et al. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016, 86, 530–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Deng, J.; Shi, T.; Chen, Q.; Liang, C.; Meng, Z.; Zhu, L.; Wang, Y.; Zhao, F.; Yu, S.; et al. Global transcriptome analysis and identification of genes involved in nutrients accumulation during seed development of rice tartary buckwheat (Fagopyrum Tararicum). Sci. Rep. 2017, 7, 11792. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ma, Z.; Zheng, T.; Sun, W.; Zhang, Y.; Jin, W.; Zhan, J.; Cai, Y.; Tang, Y.; Wu, Q.; et al. Insights into the correlation between physiological changes in and seed development of tartary buckwheat (Fagopyrum tataricum Gaertn.). BMC Genomics 2018, 19, 648. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Singh, V.K.; Rajkumar, M.S.; Kumar, V.; Jain, M. Global transcriptome and coexpression network analyses reveal cultivar-specific molecular signatures associated with seed development and seed size/weight determination in chickpea. Plant J. 2017, 91, 1088–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basnet, R.K.; Moreno-Pachon, N.; Lin, K.; Bucher, J.; Visser, R.G.; Maliepaard, C.; Bonnema, G. Genome-wide analysis of coordinated transcript abundance during seed development in different Brassica rapa morphotypes. BMC Genomics 2013, 14, 840. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.H.; Wang, H.Y.; Chen, X.; Li, Y.L.; Hussain, N.; Cui, L.B.; Wu, D.Z.; Jiang, L.X. Identification of candidate genes involved in fatty acids degradation at the late maturity stage in Brassica napus based on transcriptomic analysis. Plant Growth Regul. 2017, 83, 385–396. [Google Scholar] [CrossRef]
- Yasui, Y.; Hirakawa, H.; Ueno, M.; Matsui, K.; Katsube-Tanaka, T.; Yang, S.J.; Aii, J.; Sato, S.; Mori, M. Assembly of the draft genome of buckwheat and its applications in identifying agronomically useful genes. DNA Res. 2016, 23, 215–224. [Google Scholar] [CrossRef]
- Przybylski, R.; Gruczynska, E. A review of nutritional and nutraceutical components of buckwheat. Eur. J. Plant Sci. Biotechnol. 2009, 3, 10–22. [Google Scholar]
- Comino, I.; de Lourdes Moreno, M.; Real, A.; Rodríguez-Herrera, A.; Barro, F.; Sousa, C. The gluten-free diet: Testing alternative cereals tolerated by celiac patients. Nutrients 2013, 5, 4250–4268. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Bastida, J.A.; Zielinski, H. Buckwheat as a functional food and its effects on health. J. Agric. Food Chem. 2015, 63, 7896–7913. [Google Scholar] [CrossRef]
- Chen, Y.; Lee, C.K.; Su, F.T.; Liao, Y.L.; Chen, H.B. De Novo sequencing and characterization of developing seed transcriptome in two buckwheat species and metabolome profiling. Planta Med. 2012, 78, 11. [Google Scholar] [CrossRef]
- Gao, J.; Wang, T.; Liu, M.; Liu, J.; Zhang, Z. Transcriptome analysis of filling stage seeds among three buckwheat species with emphasis on rutin accumulation. PLoS ONE 2017, 12, e0189672. [Google Scholar] [CrossRef]
- Shi, T.X.; Li, R.Y.; Chen, Q.J.; Li, Y.; Pan, F.; Chen, Q.F. De novo sequencing of seed transcriptome and development of genic-SSR markers in common buckwheat (Fagopyrum esculentum). Mol. Breeding 2017, 37, 147. [Google Scholar] [CrossRef]
- Jain, M.; Pathak, B.P.; Harmon, A.C.; Tillman, B.L.; Gallo, M. Calcium dependent protein kinase (CDPK) expression during fruit development in cultivated peanut (Arachis hypogaea) under Ca2⁺-sufficient and -deficient growth regimens. J. Plant Physiol. 2011, 168, 2272–2277. [Google Scholar] [CrossRef]
- Pawełek, A.; Szmidt-Jaworska, A.; Świeżawska, B.; Jaworski, K. Genomic structure and promoter characterization of the CDPK kinase gene expressed during seed formation in nil. J. Plant Physiol. 2015, 189, 87–96. [Google Scholar] [CrossRef]
- Kawasaki, T.; Hayashida, N.; Baba, T.; Shinozaki, K.; Shimada, H. The gene encoding a calcium-dependent protein kinase located near the sbe1 gene encoding starch branching enzyme I is specifically expressed in developing rice seeds. Gene 1993, 129, 183–189. [Google Scholar] [CrossRef]
- Frattini, M.; Morello, L.; Breviario, D. Rice calcium-dependent protein kinase isoforms OsCDPK2 and OsCDPK11 show different response to light and different expression patterns during seed development. Plant Mol. Biol. 1999, 41, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Szczegielniak, J.; Klimecka, M.; Liwosz, A.; Ciesielski, A.; Kaczanowski, S.; Dobrowolska, G.; Harmon, A.C.; Muszyńska, G. A wound-responsive and phospholipid-regulated maize calcium-dependent protein kinase. Plant Physiol. 2005, 139, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Morello, L.; Frattini, M.; Giani, S.; Christou, P.; Breviario, D. Overexpression of the calcium-dependent protein kinase OsCDPK2 in transgenic rice is repressed by light in leaves and disrupts seed development. Transgenic Res. 2000, 9, 453–462. [Google Scholar] [CrossRef]
- Shimada, H.; Koishihara, H.; Saito, Y.; Arashima, Y.; Furukawa, T.; Hayash, H. A rice antisense SPK transformant that lacks the accumulation of seed storage substances shows no correlation between sucrose concentration in phloem sap and demand for carbon sources in the sink organs. Plant Cell Physiol. 2004, 45, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hill, A.T.; Pyc, M.; Anderson, E.M.; Snedden, W.A.; Mullen, R.T.; She, Y.M.; Plaxton, W.C. Regulatory phosphorylation of bacterial-type PEP carboxylase by the Ca2+-dependent protein kinase RcCDPK1 in developing castor oil seeds. Plant Physiol. 2017, 174, 1012–1027. [Google Scholar] [CrossRef]
- Jofuku, K.D.; Omidyar, P.K.; Gee, Z.; Okamuro, J.K. Control of seed mass and seed yield by the floral homeotic gene APETALA2. Proc. Natl. Acad. Sci. USA 2005, 102, 3117–3122. [Google Scholar] [CrossRef] [PubMed]
- Ohto, M.A.; Fischer, R.L.; Goldberg, R.B.; Nakamura, K.; Harada, J.J. Control of seed mass by APETALA2. Proc. Natl. Acad. Sci. USA 2005, 102, 3123–3128. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, L.; Corke, F.; Smith, C.; Bevan, M.W. Control of final seed and organ size by the DA1 gene family in Arabidopsis thaliana. Genes Dev. 2008, 22, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Disch, S.; Anastasiou, E.; Sharma, V.K.; Laux, T.; Fletcher, J.C.; Lenhard, M. The E3 ubiquitin ligase BIG BROTHER controls Arabidopsis organ size in a dosage-dependent manner. Curr. Biol. 2006, 16, 272–279. [Google Scholar] [CrossRef]
- Feng, Z.; Wu, C.; Wang, C.; Roh, J.; Zhang, L.; Chen, J.; Zhang, S.; Zhang, H.; Yang, C.; Hu, J.; et al. SLG controls grain size and leaf angle by modulating brassinosteroid homeostasis in rice. J. Exp. Bot. 2016, 67, 4241–4253. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Dennis, E.S.; Berger, F.; Peacock, W.J.; Chaudhury, A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc. Natl. Acad. Sci. USA 2005, 102, 17531–17536. [Google Scholar] [CrossRef] [PubMed]
- Che, R.; Tong, H.; Shi, B.; Liu, Y.; Fang, S.; Liu, D.; Xiao, Y.; Hu, B.; Liu, L.; Wang, H.; et al. Control of grain size and rice yield by GL2-mediated brassinosteroid responses. Nat. Plants 2015, 2, 15195. [Google Scholar] [CrossRef] [PubMed]
- Duan, P.; Ni, S.; Wang, J.; Zhang, B.; Xu, R.; Wang, Y.; Chen, H.; Zhu, X.; Li, Y. Regulation of OsGRF4 by OsmiR396 controls grain size and yield in rice. Nat. Plants 2016, 14, 2134–2146. [Google Scholar] [CrossRef]
- Nagasawa, N.; Hibara, K.; Heppard, E.P.; Vander Velden, K.A.; Luck, S.; Beatty, M.; Nagato, Y.; Sakai, H. GIANT EMBRYO encodes CYP78A13, required for proper size balance between embryo and endosperm in rice. Plant J. 2013, 75, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Fang, J.; Ou, S.; Gao, S.; Zhang, F.; Du, L.; Xiao, Y.; Wang, H.; Sun, X.; Chu, J.; et al. Variations in CYP78A13 coding region influence grain size and yield in rice. Plant Cell Environ. 2015, 38, 800–811. [Google Scholar] [CrossRef]
- Wu, Y.; Fu, Y.; Zhao, S.; Gu, P.; Zhu, Z.; Sun, C.; Tan, L. CLUSTERED PRIMARY BRANCH 1, a new allele of DWARF11, controls panicle architecture and seed size in rice. Plant Biotechnol. J. 2016, 14, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.B.; Huang, H.Y.; Hu, Y.W.; Zhu, S.W.; Wang, Z.Y.; Lin, W.H. Brassinosteroid regulates seed size and shape in Arabidopsis. Plant Physiol. 2013, 162, 1965–1977. [Google Scholar] [CrossRef]
- Tian, X.; Li, X.; Zhou, W.; Ren, Y.; Wang, Z.; Liu, Z.; Tang, J.; Tong, H.; Fang, J.; Bu, Q. Transcription factor OsWRKY53 positively regulates brassinosteroid signaling and plant architecture. Plant Physiol. 2017, 175, 1337–1349. [Google Scholar] [CrossRef]
- Meng, L.S.; Wang, Z.B.; Yao, S.Q.; Liu, A. The ARF2-ANT-COR15A gene cascade regulates ABA signaling-mediated resistance of large seeds to drought in Arabidopsis. J. Cell Sci. 2015, 128, 3922–3932. [Google Scholar] [CrossRef]
- Garcia, D.; Fitz Gerald, J.N.; Berger, F. Maternal control of integument cell elongation and zygotic control of endosperm growth are coordinated to determine seed size in Arabidopsis. Plant Cell 2005, 17, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Nougue, O.; Corbi, J.; Ball, S.G.; Manicacci, D.; Tenaillon, M.I. Molecular evolution accompanying functional divergence of duplicated genes along the plant starch biosynthesis pathway. BMC Evol. Biol. 2014, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI BLAST: A new generation of protein database search programs. Nucleic. Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic. Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic. Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic. Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic. Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-DELTADELTACT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | S1-1 | S1-2 | S1-3 | S2-1 | S2-2 | S2-3 | S3-1 | S3-2 | S3-3 |
|---|---|---|---|---|---|---|---|---|---|
| Raw Reads | 25,672,949 | 22,929,589 | 24,865,374 | 25,424,457 | 27,829,981 | 30,527,439 | 25,646,270 | 37,234,140 | 28,939,774 |
| Clean Reads | 25,494,672 | 22,747,870 | 24,566,718 | 25,250,033 | 27,625,287 | 30,212,779 | 21,406,245 | 36,901,842 | 28,656,479 |
| GC Content (%) | 47.77 | 48.46 | 47.65 | 48.94 | 48.82 | 49.71 | 48.61 | 50.58 | 48.33 |
| Q_30 (%) | 93.82 | 93.65 | 93.38 | 93.59 | 93.80 | 94.02 | 94.46 | 94.18 | 93.63 |
| Mapped Reads | 17,576,027 (68.94%) | 14,624,606 (64.29%) | 16,309,844 (66.39%) | 17,149,822 (67.92%) | 17,492,332 (63.32%) | 19,840,732 (65.67%) | 14,868,778 (69.46%) | 25,263,001 (68.46%) | 19,801,627 (69.21%) |
| Unique Mapped Reads | 17,327,070 (68.14%) | 14,438,073 (63.47%) | 16,088,744 (65.49%) | 16,942,772 (67.10%) | 17,265,804 (62.50%) | 19,611,115 (64.91%) | 14,671,840 (68.54%) | 25,000,998 (67.75%) | 19,580,972 (68.33%) |
| Multiple Mapped Reads | 203,957 (0.80%) | 186,533 (0.82%) | 221,100 (0.90%) | 207,050 (0.82%) | 226,528 (0.82%) | 229,617 (0.76%) | 196,938 (0.92%) | 262,003 (0.71%) | 220,655 (0.88%) |
| Gene ID | S1 vs. S2 | S2 vs. S3 | Annotation | |||||
|---|---|---|---|---|---|---|---|---|
| FDR | Log2FC | up/down | FDR | Log2FC | up/down | |||
| CaM/CML | Fes_sc0060020.1.g000001.aua.1 | 4.52 × 10−6 | 1.84 | up | 0.00005 | 1.23 | up | Calcium-like protein CML38 |
| Fes_sc0006049.1.g000004.aua.1 | - | - | - | 1.17 × 10−9 | 2.25 | up | Calmodulin-like protein 4 | |
| Fes_sc0002338.1.g000002.aua.1 | - | - | - | 3.71 × 10−14 | 1.74 | up | Calmodulin-like protein 9 | |
| CBL | Fes_sc0011071.1.g000006.aua.1 | 7.98 × 10−14 | 1.21 | up | 0.00021 | −1.89 | down | Calmodulin binding protein-like |
| Fes_sc0011071.1.g000006.aua.1 | - | - | - | 3.54 × 10−18 | −1.89 | down | Calmodulin binding protein-like | |
| CCX | Fes_sc0000003.1.g000052.aua.1 | - | - | - | 3.39 × 10−7 | −1.94 | down | Cation calcium exchanger 4 |
| Fes_sc0000035.1.g000047.aua.1 | - | - | - | 5.08 × 10−9 | −2.11 | down | Cation calcium exchanger 3 | |
| CDPK | Fes_sc0006858.1.g000001.aua.1 | 0.00016 | −3.92 | down | - | - | - | Calcium-dependent protein kinase 1 |
| Fes_sc0219194.1.g000001.aua.1 | 0.00049 | 1.56 | up | 0.00023 | −3.15 | down | Calcium-dependent protein kinase 8 | |
| Fes_sc0000035.1.g000053.aua.1 | 6.85 × 10−11 | 1.08 | up | 0.00013 | −3.33 | down | Calcium-dependent protein kinase 13 | |
| Fes_sc0080717.1.g000001.aua.1 | - | - | - | 1.83 × 10−8 | −4.95 | down | Calcium-dependent protein kinase 1 | |
| CIPK | Fes_sc0008411.1.g000003.aua.1 | 4.26 × 10−7 | 1.08 | up | 0.00024 | −2.68 | down | CBL-interacting -protein kinase |
| Fes_sc0220933.1.g000001.aua.1 | - | - | - | 3.22 × 10−6 | −3.43 | down | CBL-interacting protein kinase 18 | |
| Fes_sc0097817.1.g000001.aua.1 | - | - | - | 2.67 × 10−6 | −2.60 | down | CBL-interacting protein kinase 2 | |
| Fes_sc0093645.1.g000001.aua.1 | - | - | - | 3.39 × 10−9 | −2.31 | down | CBL-interacting protein kinase 5 | |
| Fes_sc0000542.1.g000013.aua.1 | - | - | - | 0.00004 | −2.44 | down | CBL-interacting protein kinase 7 | |
| Ca2+-ATPase | Fes_sc0074374.1.g000001.aua.1 | 1.88 × 10−11 | 1.24 | up | 0.00002 | −5.85 | down | Calcium-transporting ATPase 10 |
| Fes_sc0049771.1.g000001.aua.1 | 3.53 × 10−8 | 1.74 | up | 3.30 × 10−16 | −2.19 | down | Calcium-transporting ATPase 8 | |
| Fes_sc0023460.1.g000001.aua.1 | - | - | - | 0.00009 | −2.64 | down | Calcium-transporting ATPase 1 | |
| Fes_sc0009288.1.g000009.aua.1 | - | - | - | 0.00026 | −2.00 | down | Calcium-transporting ATPase 10 | |
| CHX | Fes_sc0150773.1.g000001.aua.1 | - | - | - | 0.00008 | −7.37 | down | Cation/H(+) antiporter 17 |
| Fesculentum_newGene_621 | - | - | - | 5.17 × 10−13 | −4.99 | down | Cation/H(+) antiporter 15 | |
| Gene ID | S1 vs. S2 | S2 vs. S3 | Annotation | |||||
|---|---|---|---|---|---|---|---|---|
| FDR | Log2FC | up/down | FDR | Log2FC | up/down | |||
| Auxin | Fes_sc0003131.1.g000008.aua.1 | 3.57 × 10−7 | 2.13 | up | - | - | - | Auxin responsive protein/IAA |
| Fes_sc0008308.1.g000001.aua.1 | - | - | - | 0.00023 | −1.73 | down | Auxin-responsive protein/IAA12 | |
| Fes_sc0076315.1.g000001.aua.1 | - | - | - | 0.00239 | −5.90 | down | Auxin transporter-like protein/AUX1 | |
| Fes_sc0096151.1.g000001.aua.1 | - | - | - | 4.49 × 10−14 | −3.12 | down | Auxin-responsive protein/IAA9 | |
| Fes_sc0032547.1.g000002.aua.1 | 1.85 × 10−6 | 2.08 | up | 0.00626 | 1.76 | up | Auxin response factor 7/ARF7 | |
| Fes_sc0001323.1.g000012.aua.1 | - | - | - | 0.00042 | 4.27 | up | Auxin responsive protein/IAA | |
| Fes_sc0006670.1.g000009.aua.1 | - | - | - | 3.41 × 10−9 | 2.07 | up | Auxin-induced protein/IAA | |
| Fes_sc0007310.1.g000002.aua.1 | - | - | - | 1.36 × 10−15 | 3.66 | up | Auxin responsive protein/IAA | |
| Fes_sc0007969.1.g000004.aua.1 | - | - | - | 1.90 × 10−8 | 2.19 | up | Auxin responsive protein/IAA | |
| Fes_sc0010815.1.g000005.aua.1 | - | - | - | 7.27 × 10−14 | 2.26 | up | Auxin responsive protein/IAA | |
| Cytokinine | Fes_sc0001025.1.g000012.aua.1 | - | - | - | 0.00019 | 1.93 | up | Histidine-containing phosphotransfer protein 1/AHP |
| Fes_sc0040103.1.g000001.aua.1 | 0.028 | 3.44 | up | - | - | - | Two-component response regulator /ARR8 | |
| Gibberellin | Fes_sc0005307.1.g000002.aua.1 | 5.01 × 10−18 | 1.22 | up | 0.00011 | 1.47 | up | Transcription factor/PIF3 |
| Fes_sc0000054.1.g000047.aua.1 | - | - | - | 2.56 × 10−6 | 4.92 | up | Transcription factor bHLH127 | |
| Abscisic acid | Fes_sc0011976.1.g000004.aua.1 | - | - | - | 0.00118 | −2.93 | down | Abscisic acid receptor/PYR/PYL |
| Fes_sc0002839.1.g000004.aua.1 | 2.99 × 10−15 | 2.38 | up | Protein phosphatase 2C/PP2C | ||||
| Fes_sc0011132.1.g000002.aua.1 | 0.0001 | 2.17 | up | Protein phosphatase 2C/PP2C | ||||
| Fes_sc0000642.1.g000010.aua.1 | - | - | - | 4.93 × 10−10 | −1.72 | down | Protein phosphatase 2C/PP2C | |
| Fes_sc0002743.1.g000004.aua.1 | - | - | - | 2.60 × 10−7 | −1.83 | down | Serine/threonine-protein kinase/SRK2A | |
| Fes_sc0000024.1.g000035.aua.1 | 0.00035 | −1.93 | down | - | - | - | ABSCISIC ACID-INSENSITIVE 5 /ABF | |
| Ethylene | Fes_sc0005120.1.g000005.aua.1 | - | - | - | 4.22 × 10−15 | −3.46 | down | Protein EIN4 |
| Fes_sc0125064.1.g000001.aua.1 | - | - | - | 4.90 × 10−10 | −3.45 | down | Ethylene receptor 2/ETR | |
| Fes_sc0043049.1.g000001.aua.1 | - | - | - | 1.91 × 10−7 | −2.07 | down | Ethylene receptor 1/ETR | |
| Fes_sc0027826.1.g000001.aua.1 | - | - | - | 0.00006 | −4.68 | down | Serine/threonine-protein kinase/CTR1 | |
| Fes_sc0004642.1.g000013.aua.1 | 6.60 × 10−9 | 3.43 | up | Ethylene insensitive 3/EIN3 | ||||
| Fes_sc0006207.1.g000001.aua.1 | - | - | - | 0.03248 | 2.01 | up | Ethylene insensitive 3-like protein/EIN3 | |
| Brassinosteroid | Fes_sc0009187.1.g000001.aua.1 | 2.80 × 10−12 | 2.02 | up | 0.01759 | −2.33 | down | BRASSINOSTEROID INSENSITIVE 1/BRI1 |
| Fes_sc0361928.1.g000001.aua.1 | - | - | - | 3.83 × 10−7 | −3.60 | down | BRASSINOSTEROID INSENSITIVE 1/BRI1 | |
| Jasmonic acid | Fes_sc0000770.1.g000005.aua.1 | - | - | - | 0.00011 | 2.07 | up | Protein TIFY 6B |
| Salicylic acid | Fes_sc0002899.1.g000004.aua.1 | 0.00027 | −1.76 | down | - | - | - | bZIP transcription factor |
| Gene ID | S1 vs. S2 | S2 vs. S3 | Annotation | |||||
|---|---|---|---|---|---|---|---|---|
| FDR | Log2FC | up/down | FDR | Log2FC | up/down | |||
| SUS | Fes_sc0086881.1.g000001.aua.1 | 1.06 × 10−9 | 1.74 | up | 3.08 × 10−14 | −1.66 | down | Sucrose synthase 2 |
| Fes_sc0023486.1.g000001.aua.1 | 2.69 × 10−6 | 1.75 | up | 5.57 × 10−9 | −2.47 | down | Sucrose synthase 3 isoform 4 | |
| Fes_sc0052588.1.g000001.aua.1 | 0.00015 | 1.73 | up | - | - | - | Sucrose synthase 4 | |
| Fes_sc0053143.1.g000001.aua.1 | 0.00627 | 1.57 | up | - | - | - | Sucrose synthase 4 | |
| Fes_sc0007558.1.g000005.aua.1 | 4.33 × 10−14 | 1.51 | up | - | - | - | Sucrose synthase 3 | |
| Fes_sc0006080.1.g000001.aua.1 | 0.00001 | 1.72 | up | - | - | - | Sucrose synthase 1 | |
| Fes_sc0000045.1.g000030.aua.1 | - | - | - | 0.00035 | −1.87 | down | Sucrose synthase | |
| UGPase | Fes_sc0005131.1.g000007.aua.1 | 3.89 × 10−8 | 1.33 | up | - | - | - | UDP glucose pyrophosphorylase |
| AGPase | Fes_sc0000081.1.g000017.aua.1 | - | - | - | 1.42 × 10−6 | −2.09 | down | ADP-glucose pyrophosphorylase |
| GBSS | Fes_sc0002521.1.g000007.aua.1 | - | - | - | 0.00001 | −2.75 | down | Granule-bound starch synthase 1 |
| SS | Fes_sc0005785.1.g000003.aua.1 | 2.02 × 10−11 | 2.13 | up | 2.93 × 10−10 | −1.89 | down | Starch synthase 1 |
| Fes_sc0069832.1.g000001.aua.1 | - | - | - | 1.87 × 10−6 | −3.03 | down | Starch synthase 3 | |
| SBE | Fes_sc0000127.1.g000022.aua.1 | 0.00027 | 1.38 | up | 1.13 × 10-12 | −1.32 | down | Starch-branching enzyme |
| Fes_sc0001814.1.g000004.gia.1 | 0.00268 | 1.37 | up | - | - | - | Starch-branching enzyme | |
| DBE | Fes_sc0001905.1.g000005.aua.1 | 3.21 × 10−12 | −1.53 | up | 4.66 × 10−12 | 2.21 | down | Debranching enzyme |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Lv, Q.; Deng, J.; Huang, J.; Cai, F.; Liang, C.; Chen, Q.; Wang, Y.; Zhu, L.; Zhang, X.; et al. Transcriptome Analysis Reveals Key Seed-Development Genes in Common Buckwheat (Fagopyrum esculentum). Int. J. Mol. Sci. 2019, 20, 4303. https://doi.org/10.3390/ijms20174303
Li H, Lv Q, Deng J, Huang J, Cai F, Liang C, Chen Q, Wang Y, Zhu L, Zhang X, et al. Transcriptome Analysis Reveals Key Seed-Development Genes in Common Buckwheat (Fagopyrum esculentum). International Journal of Molecular Sciences. 2019; 20(17):4303. https://doi.org/10.3390/ijms20174303
Chicago/Turabian StyleLi, Hongyou, Qiuyu Lv, Jiao Deng, Juan Huang, Fang Cai, Chenggang Liang, Qijiao Chen, Yan Wang, Liwei Zhu, Xiaona Zhang, and et al. 2019. "Transcriptome Analysis Reveals Key Seed-Development Genes in Common Buckwheat (Fagopyrum esculentum)" International Journal of Molecular Sciences 20, no. 17: 4303. https://doi.org/10.3390/ijms20174303
APA StyleLi, H., Lv, Q., Deng, J., Huang, J., Cai, F., Liang, C., Chen, Q., Wang, Y., Zhu, L., Zhang, X., & Chen, Q. (2019). Transcriptome Analysis Reveals Key Seed-Development Genes in Common Buckwheat (Fagopyrum esculentum). International Journal of Molecular Sciences, 20(17), 4303. https://doi.org/10.3390/ijms20174303