Multi-Staged Regulation of Lipid Signaling Mediators during Myogenesis by COX-1/2 Pathways


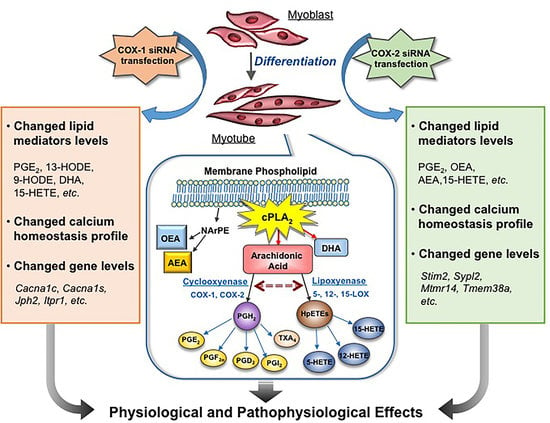
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
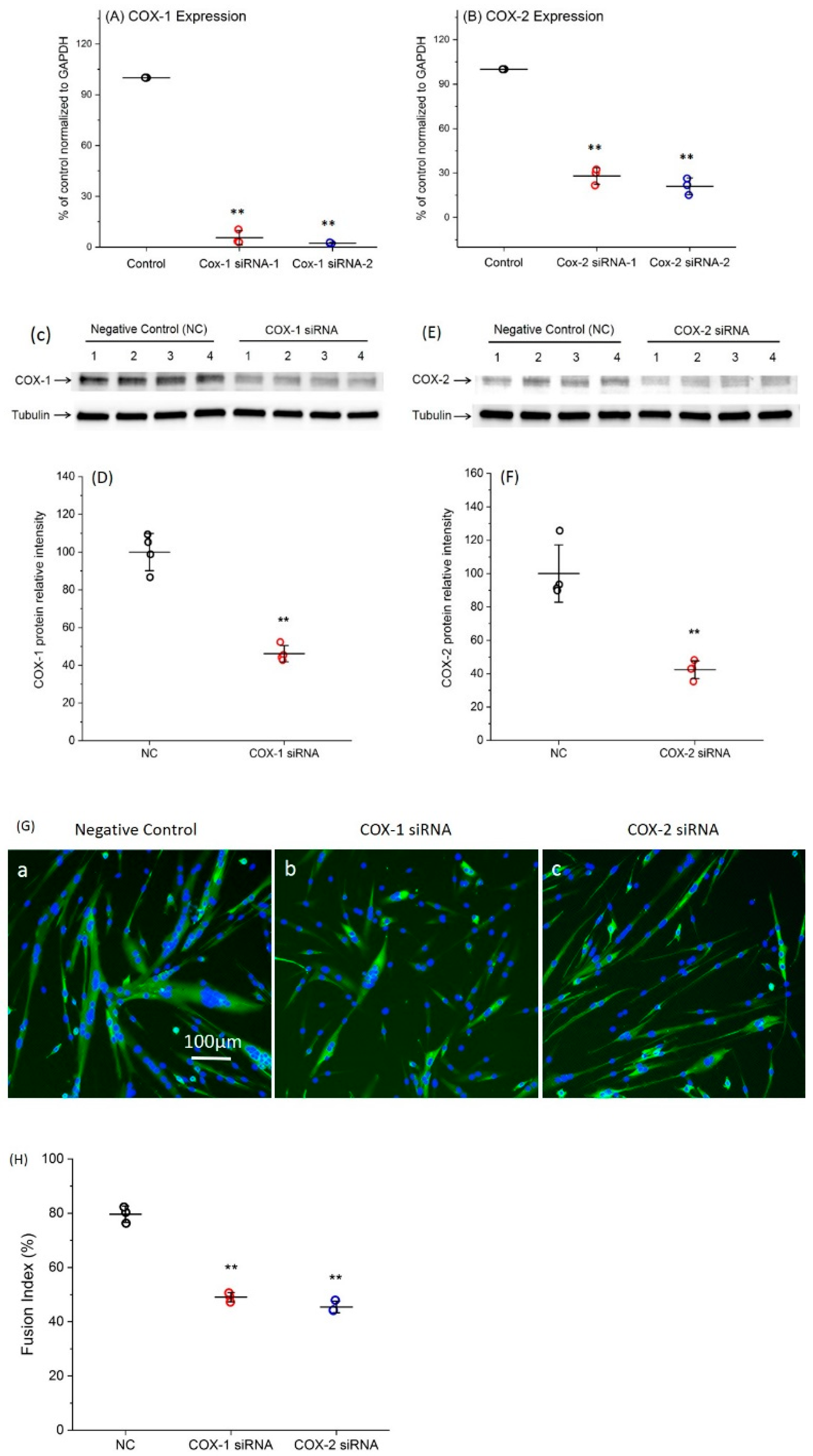
2.1. Transfection with Specific siRNAs Targeting COX-1 or -2 Significantly Reduce the Expression Levels of COXs
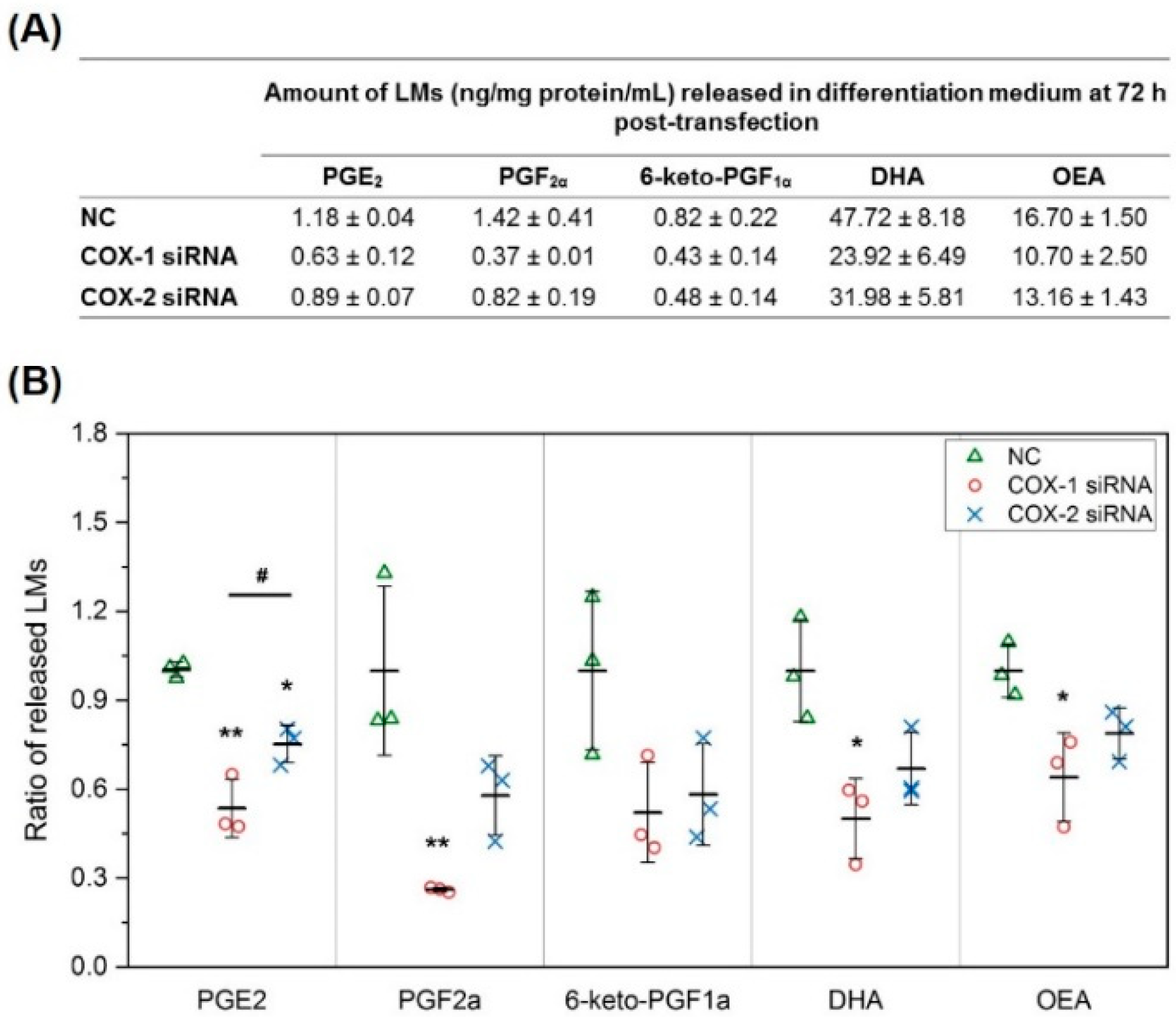
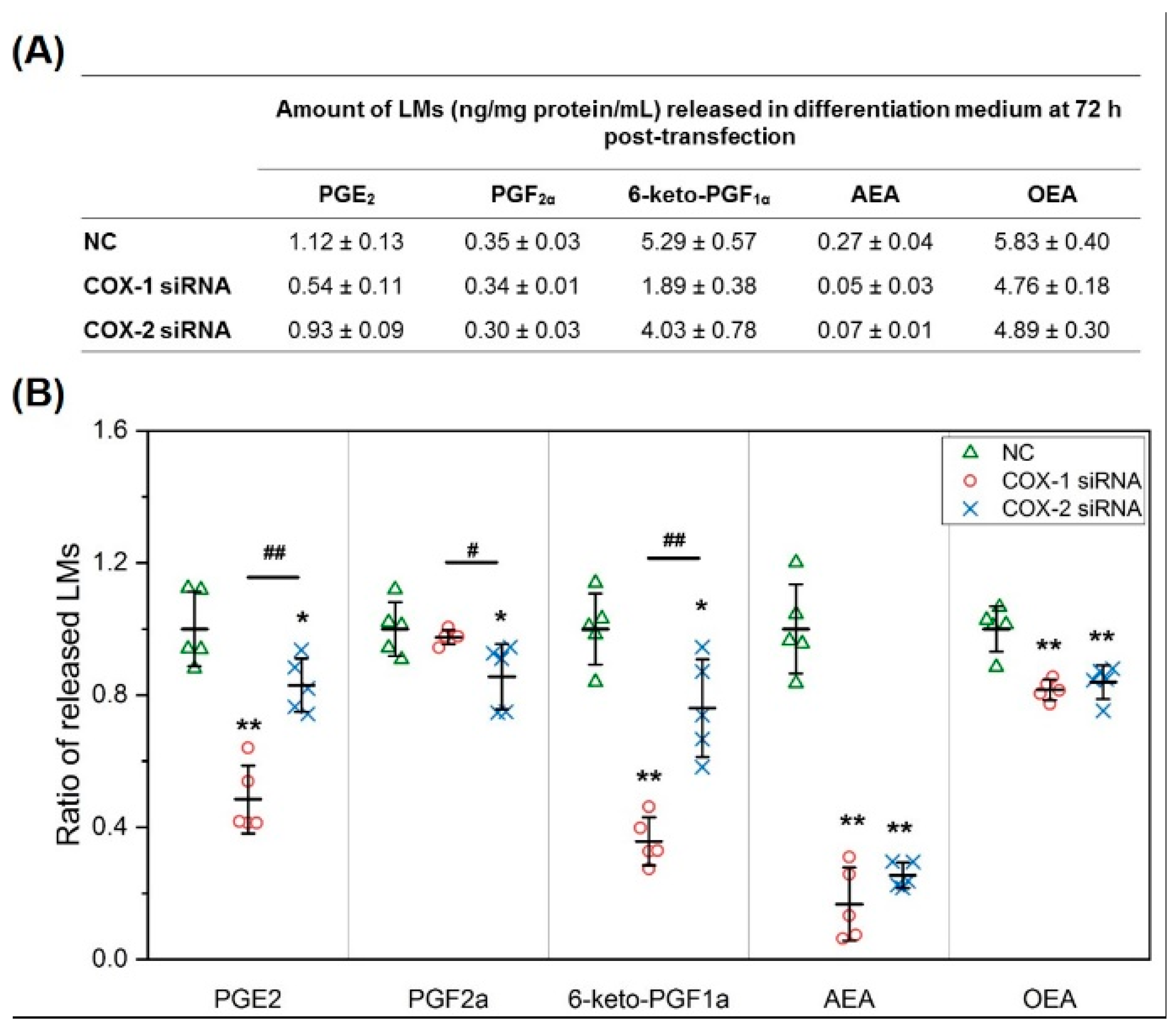
2.2. The Changes in Levels of Lipid Mediators after Knocking Down COX-1 or -2 Are not Limited to PGs and Thromboxane B2 (TXB2)
2.3. COXs could Interact with LOXs to Regulate the Levels of Lipid Mediators
2.4. Supplement with LMs Improves Defective Myogenic Differentiation of Primary Myoblast Caused by Knocking Down COX-1 or -2
2.5. Results of Lipidomic Analysis of C2C12 Cells Show Similar Patterns as Primary Myoblasts
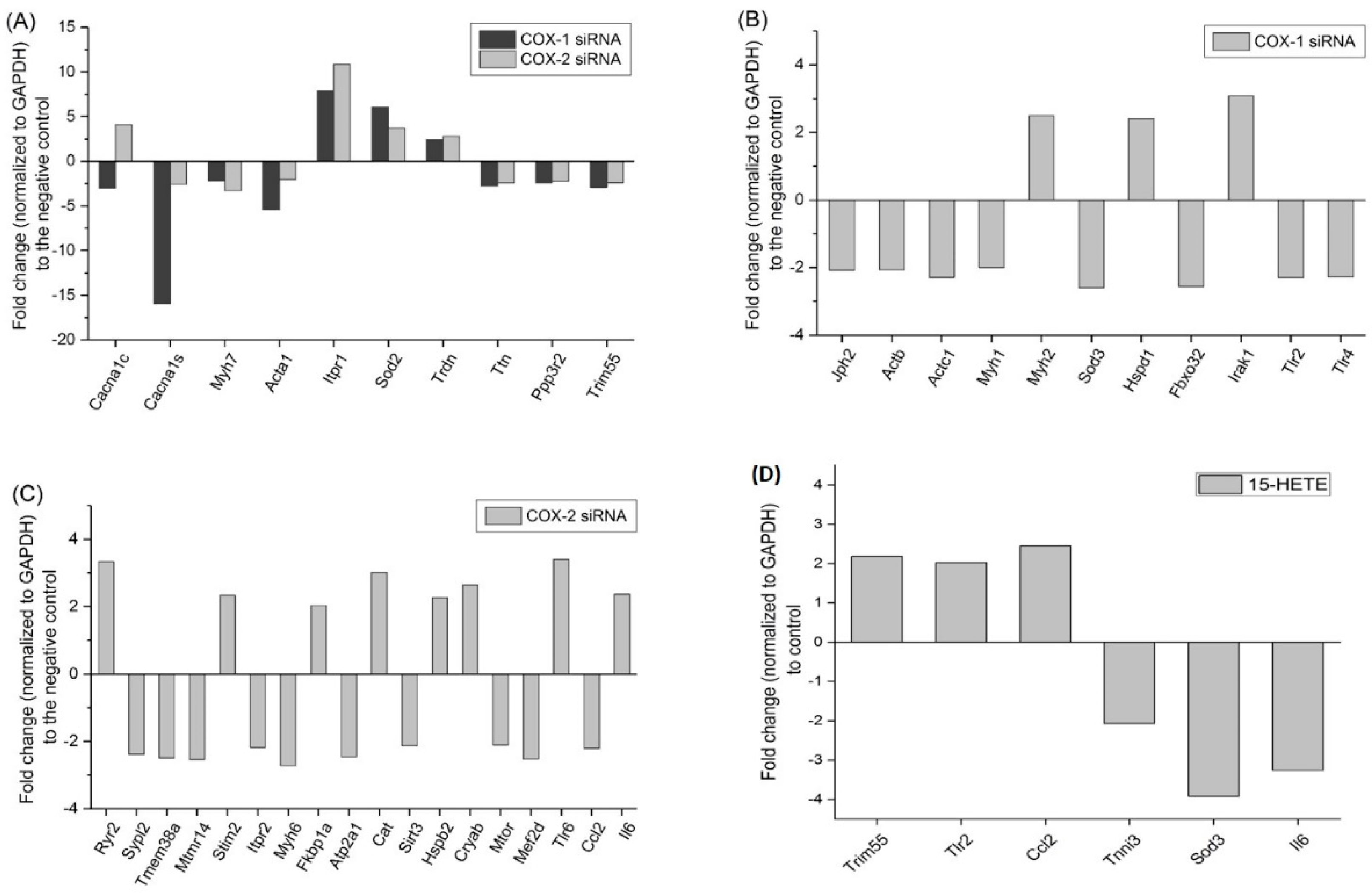
2.6. Changes in Gene Expression Profile after siRNA Transfection Targeting at COX-1 or -2
2.7. Intracellular Calcium Homeostasis Measurement
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.1.1. Myoblast Isolation and Culture
4.1.2. C2C12 Cells
4.2. siRNA Transfection
4.3. Quantitative Real-Time PCR (qRT-PCR)
4.4. Protein Sample Preparation and Western Blotting
4.5. Immunohistochemistry
4.6. LC-MS/MS
4.6.1. Sample Preparation for Lipidomics Analysis
4.6.2. LC-MS/MS Conditions
4.7. Intracellular Ca2+ Measurements
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karalaki, M.; Fili, S.; Philippou, A.; Koutsilieris, M. Muscle regeneration: Cellular and molecular events. In Vivo 2009, 23, 779–796. [Google Scholar] [PubMed]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Inazumi, T.; Tsuchiya, S. Roles of prostaglandin receptors in female reproduction. J. Biochem. 2015, 157, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Romero-Suarez, S.; Bonewald, L.; Johnson, M.; Brotto, M. Prostaglandin E2: From clinical applications to its potential role in bone—Muscle crosstalk and myogenic differentiation. Recent Pat. Biotechnol. 2012, 6, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Zhao, R.; Vallejo, J.; Igwe, O.; Bonewald, L.; Wetmore, L.; Brotto, M. Prostaglandin E2 promotes proliferation of skeletal muscle myoblasts via EP4 receptor activation. Cell Cycle 2015, 14, 1507–1516. [Google Scholar] [CrossRef]
- Jansen, K.M.; Pavlath, G.K. Prostaglandin F2alpha promotes muscle cell survival and growth through upregulation of the inhibitor of apoptosis protein BRUCE. Cell Death Differ. 2008, 15, 1619–1628. [Google Scholar] [CrossRef][Green Version]
- Horsley, V.; Pavlath, G.K. Prostaglandin F2(α) stimulates growth of skeletal muscle cells via an NFATC2-dependent pathway. J. Cell Biol. 2003, 161, 111–118. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Cyclooxygenases: Structural and functional insights. J. Lipid. Res. 2009, 50, S29–S34. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Prisk, V.; Li, Y.; Foster, W.; Huard, J. Inhibited skeletal muscle healing in cyclooxygenase-2 gene-deficient mice: The role of PGE2 and PGF2alpha. J. Appl. Physiol. 2006, 101, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Bondesen, B.A.; Mills, S.T.; Kegley, K.M.; Pavlath, G.K. The COX-2 pathway is essential during early stages of skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 2004, 287, C475–C483. [Google Scholar] [CrossRef] [PubMed]
- Bondesen, B.A.; Mills, S.T.; Pavlath, G.K. The COX-2 pathway regulates growth of atrophied muscle via multiple mechanisms. Am. J. Physiol. Cell Physiol. 2006, 290, C1651–C1659. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Z.; Hennig, R.; Adrian, T.E. Lipoxygenase and cyclooxygenase metabolism: New insights in treatment and chemoprevention of pancreatic cancer. Mol. Cancer 2003, 2, 10. [Google Scholar] [CrossRef]
- Kroetz, D.L.; Zeldin, D.C. Cytochrome P450 pathways of arachidonic acid metabolism. Curr. Opin. Lipidol. 2002, 13, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Tandon, R.; Dastidar, S.G.; Ray, A. A review on leukotrienes and their receptors with reference to asthma. J. Asthma 2013, 50, 922–931. [Google Scholar] [CrossRef]
- Moreno, J.J. New aspects of the role of hydroxyeicosatetraenoic acids in cell growth and cancer development. Biochem. Pharmcol. 2009, 77, 1–10. [Google Scholar] [CrossRef]
- Brenneis, C.; Maier, T.J.; Schmidt, R.; Hofacker, A.; Zulauf, L.; Jakobsson, P.J.; Scholich, K.; Geisslinger, G. Inhibition of prostaglandin E2 synthesis by SC-560 is independent of cyclooxygenase 1 inhibition. FASEB J. 2006, 20, 1352–1360. [Google Scholar] [CrossRef]
- Paiotti, A.P.; Marchi, P.; Miszputen, S.J.; Oshima, C.T.; Franco, M.; Ribeiro, D.A. The role of nonsteroidal antiinflammatory drugs and cyclooxygenase-2 inhibitors on experimental colitis. In Vivo 2012, 26, 381–393. [Google Scholar]
- Daikoku, T.; Wang, D.; Tranguch, S.; Morrow, J.D.; Orsulic, S.; DuBois, R.N.; Dey, S.K. Cyclooxygenase-1 is a potential target for prevention and treatment of ovarian epithelial cancer. Cancer Res. 2005, 65, 3735–3744. [Google Scholar] [CrossRef] [PubMed]
- Otis, J.S.; Burkholder, T.J.; Pavlath, G.K. Stretch-induced myoblast proliferation is dependent on the COX2 pathway. Exp. Cell Res. 2005, 310, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Testa, M.; Rocca, B.; Spath, L.; Ranelletti, F.O.; Petrucci, G.; Ciabattoni, G.; Naro, F.; Schiaffino, S.; Volpe, M.; Reggiani, C. Expression and activity of cyclooxygenase isoforms in skeletal muscles and myocardium of humans and rodents. J. Appl. Physiol. 2007, 103, 1412–1418. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Novak, M.L.; Billich, W.; Smith, S.M.; Sukhija, K.B.; McLoughlin, T.J.; Hornberger, T.A.; Koh, T.J. COX-2 inhibitor reduces skeletal muscle hypertrophy in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R1132–R1139. [Google Scholar] [CrossRef] [PubMed]
- Bondesen, B.A.; Jones, K.A.; Glasgow, W.C.; Pavlath, G.K. Inhibition of myoblast migration by prostacyclin is associated with enhanced cell fusion. FASEB J. 2007, 21, 3338–3345. [Google Scholar] [CrossRef] [PubMed]
- Velica, P.; Khanim, F.L.; Bunce, C.M. Prostaglandin D2 inhibits C2C12 myogenesis. Mol. Cell. Endocrinol. 2010, 319, 71–78. [Google Scholar] [CrossRef]
- Sears, D.D.; Miles, P.D.; Chapman, J.; Ofrecio, J.M.; Almazan, F.; Thapar, D.; Miller, Y.I. 12/15-lipoxygenase is required for the early onset of high fat diet-induced adipose tissue inflammation and insulin resistance in mice. PLoS ONE 2009, 4, e7250. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Hamilton, R.; Jernigan, A.; Zhang, Y.; Sabia, M.; Rahman, M.M.; Li, Y.; Wei, R.; Chaudhuri, A.; Van Remmen, H. Genetic ablation of 12/15-lipoxygenase but not 5-lipoxygenase protects against denervation-induced muscle atrophy. Free Radic. Biol. Med. 2014, 67, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Kitase, Y.; Vallejo, J.A.; Gutheil, W.; Vemula, H.; Jahn, K.; Yi, J.; Zhou, J.; Brotto, M.; Bonewald, L.F. β-aminoisobutyric Acid, l-BAIBA, Is a Muscle-Derived Osteocyte Survival Factor. Cell Rep. 2018, 22, 1531–1544. [Google Scholar] [CrossRef]
- Roberts, L.D.; Bostrom, P.; O’Sullivan, J.F.; Schinzel, R.T.; Lewis, G.D.; Dejam, A.; Lee, Y.K.; Palma, M.J.; Calhoun, S.; Georgiadi, A.; et al. β-Aminoisobutyric acid induces browning of white fat and hepatic β-oxidation and is inversely correlated with cardiometabolic risk factors. Cell Metab. 2014, 19, 96–108. [Google Scholar] [CrossRef]
- Lauffer, L.M.; Iakoubov, R.; Brubaker, P.L. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes 2009, 58, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Miyares, R.L.; Ahern, G.P. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J. Physiol. 2005, 564 Pt 2, 541–547. [Google Scholar] [CrossRef]
- Fu, J.; Gaetani, S.; Oveisi, F.; Lo Verme, J.; Serrano, A.; Rodriguez De Fonseca, F.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G.; et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Lo Verme, J.; Fu, J.; Oveisi, F.; Blazquez, C.; Piomelli, D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-α). J. Biol. Chem. 2004, 279, 27849–27854. [Google Scholar] [CrossRef] [PubMed]
- Bowen, K.J.; Kris-Etherton, P.M.; Shearer, G.C.; West, S.G.; Reddivari, L.; Jones, P.J.H. Oleic acid-derived oleoylethanolamide: A nutritional science perspective. Prog. Lipid Res. 2017, 67, 1–15. [Google Scholar] [CrossRef]
- Pillarisetti, S.; Alexander, C.W.; Khanna, I. Pain and beyond: Fatty acid amides and fatty acid amide hydrolase inhibitors in cardiovascular and metabolic diseases. Drug Discov. Today 2009, 14, 1098–1111. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Huenchullan, S.; McLennan, S.V.; Verhoeven, A.; Twigg, S.M.; Tam, C.S. The emerging role of skeletal muscle extracellular matrix remodelling in obesity and exercise. Obes. Rev. 2017, 18, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Gurkoff, G.; Shahlaie, K.; Lyeth, B.; Berman, R. Voltage-gated calcium channel antagonists and traumatic brain injury. Pharmaceuticals (Basel) 2013, 6, 788–812. [Google Scholar] [CrossRef]
- Shen, J.; Yu, W.M.; Brotto, M.; Scherman, J.A.; Guo, C.; Stoddard, C.; Nosek, T.M.; Valdivia, H.H.; Qu, C.K. Deficiency of MIP/MTMR14 phosphatase induces a muscle disorder by disrupting Ca2+ homeostasis. Nat. Cell Biol. 2009, 11, 769–776. [Google Scholar] [CrossRef]
- Weisleder, N.; Brotto, M.; Komazaki, S.; Pan, Z.; Zhao, X.; Nosek, T.; Parness, J.; Takeshima, H.; Ma, J. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J. Cell Biol. 2006, 174, 639–645. [Google Scholar] [CrossRef]
- Yazawa, M.; Ferrante, C.; Feng, J.; Mio, K.; Ogura, T.; Zhang, M.; Lin, P.H.; Pan, Z.; Komazaki, S.; Kato, K.; et al. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature 2007, 448, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.; Holt, M.R.; Mankoo, B.S.; Gautel, M. Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Dev. Biol. 2011, 351, 46–61. [Google Scholar] [CrossRef] [PubMed]
- McElhinny, A.S.; Perry, C.N.; Witt, C.C.; Labeit, S.; Gregorio, C.C. Muscle-specific RING finger-2 (MURF-2) is important for microtubule, intermediate filament and sarcomeric M-line maintenance in striated muscle development. J. Cell Sci. 2004, 117 Pt 15, 3175–3188. [Google Scholar] [CrossRef]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The Role of TLR2 in Infection and Immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [PubMed]
- Mojumdar, K.; Giordano, C.; Lemaire, C.; Liang, F.; Divangahi, M.; Qureshi, S.T.; Petrof, B.J. Divergent impact of Toll-like receptor 2 deficiency on repair mechanisms in healthy muscle versus Duchenne muscular dystrophy. J. Pathol. 2016, 239, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Zbinden-Foncea, H.; Raymackers, J.M.; Deldicque, L.; Renard, P.; Francaux, M. TLR2 and TLR4 activate p38 MAPK and JNK during endurance exercise in skeletal muscle. Med. Sci Sports Exerc. 2012, 44, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Cha, H.N.; Jo, H.J.; Song, I.H.; Baek, S.H.; Dan, J.M.; Kim, Y.W.; Kim, J.Y.; Lee, I.K.; Seo, J.S.; et al. TLR2 deficiency attenuates skeletal muscle atrophy in mice. Biochem. Biophys. Res. Commun. 2015, 459, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Divangahi, M.; Yahiaoui, L.; Gvozdic, D.; Qureshi, S.; Petrof, B.J. Toll-like receptors differentially regulate CC and CXC chemokines in skeletal muscle via NF-κB and calcineurin. Infect. Immun. 2006, 74, 6829–6838. [Google Scholar] [CrossRef] [PubMed]
- Hubal, M.J.; Devaney, J.M.; Hoffman, E.P.; Zambraski, E.J.; Gordish-Dressman, H.; Kearns, A.K.; Larkin, J.S.; Adham, K.; Patel, R.R.; Clarkson, P.M. CCL2 and CCR2 polymorphisms are associated with markers of exercise-induced skeletal muscle damage. J. Appl. Physiol. 2010, 108, 1651–1658. [Google Scholar] [CrossRef]
- Harmon, B.T.; Orkunoglu-Suer, E.F.; Adham, K.; Larkin, J.S.; Gordish-Dressman, H.; Clarkson, P.M.; Thompson, P.D.; Angelopoulos, T.J.; Gordon, P.M.; Moyna, N.M.; et al. CCL2 and CCR2 variants are associated with skeletal muscle strength and change in strength with resistance training. J. Appl. Physiol. 2010, 109, 1779–1785. [Google Scholar] [CrossRef][Green Version]
- Lu, H.; Huang, D.; Ransohoff, R.M.; Zhou, L. Acute skeletal muscle injury: CCL2 expression by both monocytes and injured muscle is required for repair. FASEB J. 2011, 25, 3344–3355. [Google Scholar] [CrossRef] [PubMed]
- Hindi, S.M.; Kumar, A. Toll-like receptor signalling in regenerative myogenesis: Friend and foe. J. Pathol. 2016, 239, 125–128. [Google Scholar] [CrossRef]
- Cantini, M.; Massimino, M.L.; Rapizzi, E.; Rossini, K.; Catani, C.; Dalla Libera, L.; Carraro, U. Human satellite cell proliferation in vitro is regulated by autocrine secretion of IL-6 stimulated by a soluble factor(s) released by activated monocytes. Biochem. Biophys. Res. Commun. 1995, 216, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Yakabe, M.; Ogawa, S.; Ota, H.; Iijima, K.; Eto, M.; Ouchi, Y.; Akishita, M. Inhibition of interleukin-6 decreases atrogene expression and ameliorates tail suspension-induced skeletal muscle atrophy. PLoS ONE 2018, 13, e0191318. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hsu, Y.H.; Mo, C.; Abreu, E.; Kiel, D.P.; Bonewald, L.F.; Brotto, M.; Karasik, D. METTL21C is a potential pleiotropic gene for osteoporosis and sarcopenia acting through the modulation of the NF-κB signaling pathway. J. Bone Min. Res. 2014, 29, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Bian, L.; Mo, C.; Kukula, M.; Schug, K.A.; Brotto, M. Targeted quantification of lipid mediators in skeletal muscles using restricted access media-based trap-and-elute liquid chromatography-mass spectrometry. Anal. Chim. Acta 2017, 984, 151–161. [Google Scholar] [CrossRef]
- Huang, J.; Romero-Suarez, S.; Lara, N.; Mo, C.; Kaja, S.; Brotto, L.; Dallas, S.L.; Johnson, M.L.; Jähn, K.; Bonewald, L.F.; et al. Crosstalk between MLO-Y4 osteocytes and C2C12 muscle cells is mediated by the Wnt/β-catenin pathway. JBMR Plus 2017, 1, 86–100. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mo, C.; Wang, Z.; Bonewald, L.; Brotto, M. Multi-Staged Regulation of Lipid Signaling Mediators during Myogenesis by COX-1/2 Pathways. Int. J. Mol. Sci. 2019, 20, 4326. https://doi.org/10.3390/ijms20184326
Mo C, Wang Z, Bonewald L, Brotto M. Multi-Staged Regulation of Lipid Signaling Mediators during Myogenesis by COX-1/2 Pathways. International Journal of Molecular Sciences. 2019; 20(18):4326. https://doi.org/10.3390/ijms20184326
Chicago/Turabian StyleMo, Chenglin, Zhiying Wang, Lynda Bonewald, and Marco Brotto. 2019. "Multi-Staged Regulation of Lipid Signaling Mediators during Myogenesis by COX-1/2 Pathways" International Journal of Molecular Sciences 20, no. 18: 4326. https://doi.org/10.3390/ijms20184326
APA StyleMo, C., Wang, Z., Bonewald, L., & Brotto, M. (2019). Multi-Staged Regulation of Lipid Signaling Mediators during Myogenesis by COX-1/2 Pathways. International Journal of Molecular Sciences, 20(18), 4326. https://doi.org/10.3390/ijms20184326