The Role of Lysosomes in a Broad Disease-Modifying Approach Evaluated across Transgenic Mouse Models of Alzheimer’s Disease and Parkinson’s Disease and Models of Mild Cognitive Impairment

 ,
, 
Abstract
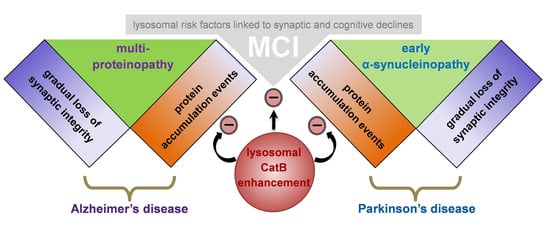
:
1. Introduction
2. Results
2.1. Assessment of Protein Accumulation Pathology in the APP/PS1 Transgenic Mouse Model of AD
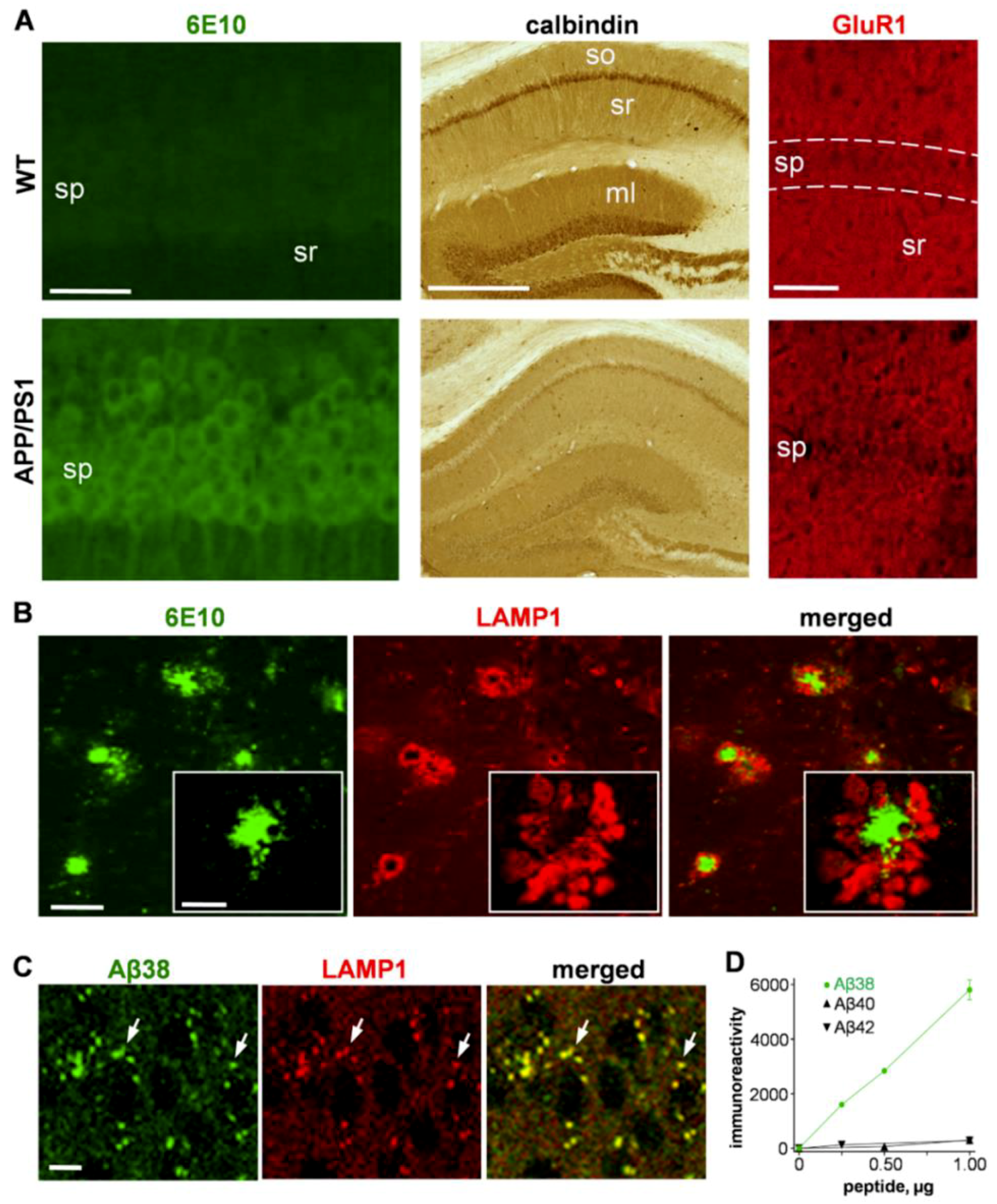
2.1.1. Immunostaining Aβ Species, Synaptic Markers, and Lysosomal-Associated Membrane Protein 1 (LAMP1)
2.1.2. Colocalization of LAMP1 and the Aβ42 Degradation Product Aβ38 in Neurons
2.2. Positive CatB Modulation as a Compensatory Strategy in APP/PS1 Transgenic Mice
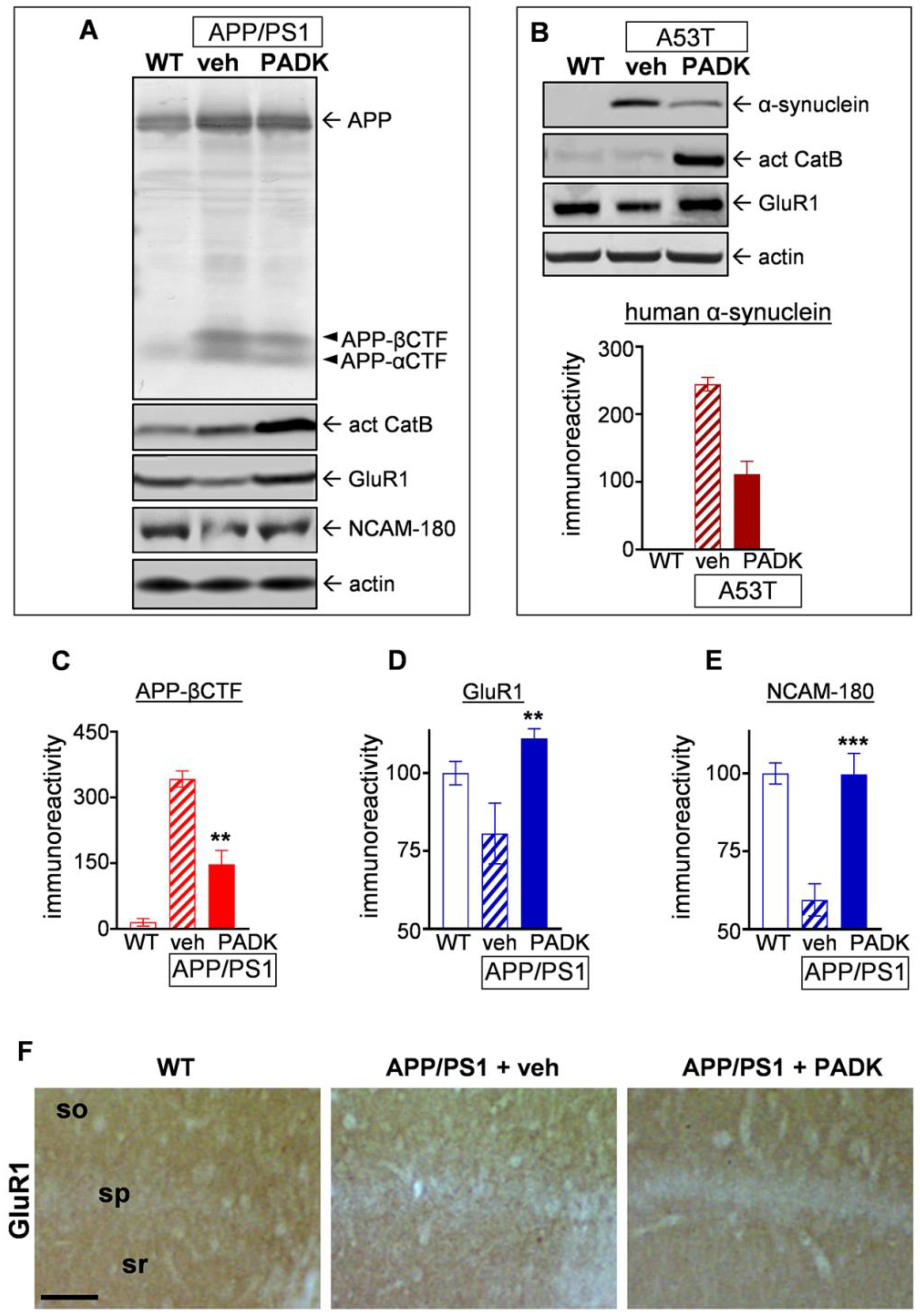
2.2.1. CatB Enhancement by Z-Phe-Ala-Diazomethylketone (PADK) is Associated with Improved Aβ42 Detoxification in Lysosomes
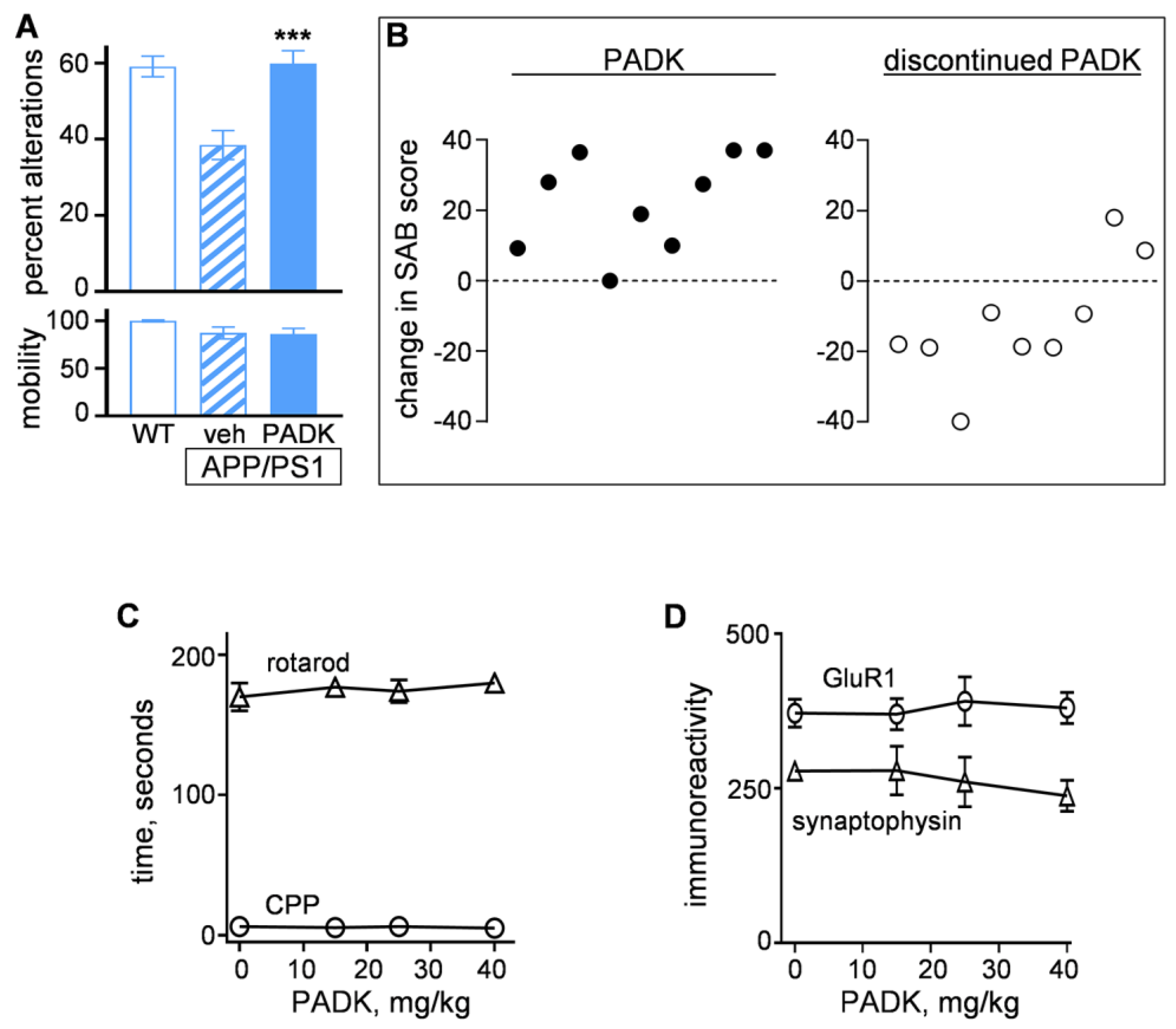
2.2.2. Positive CatB Modulator Reduces Protein Accumulation Events and Improves Synaptic and Cognitive Measures
2.3. Assessment of Positive CatB Modulation as a Potential Preventive Strategy in Models of Age-Related Cognitive Impairment
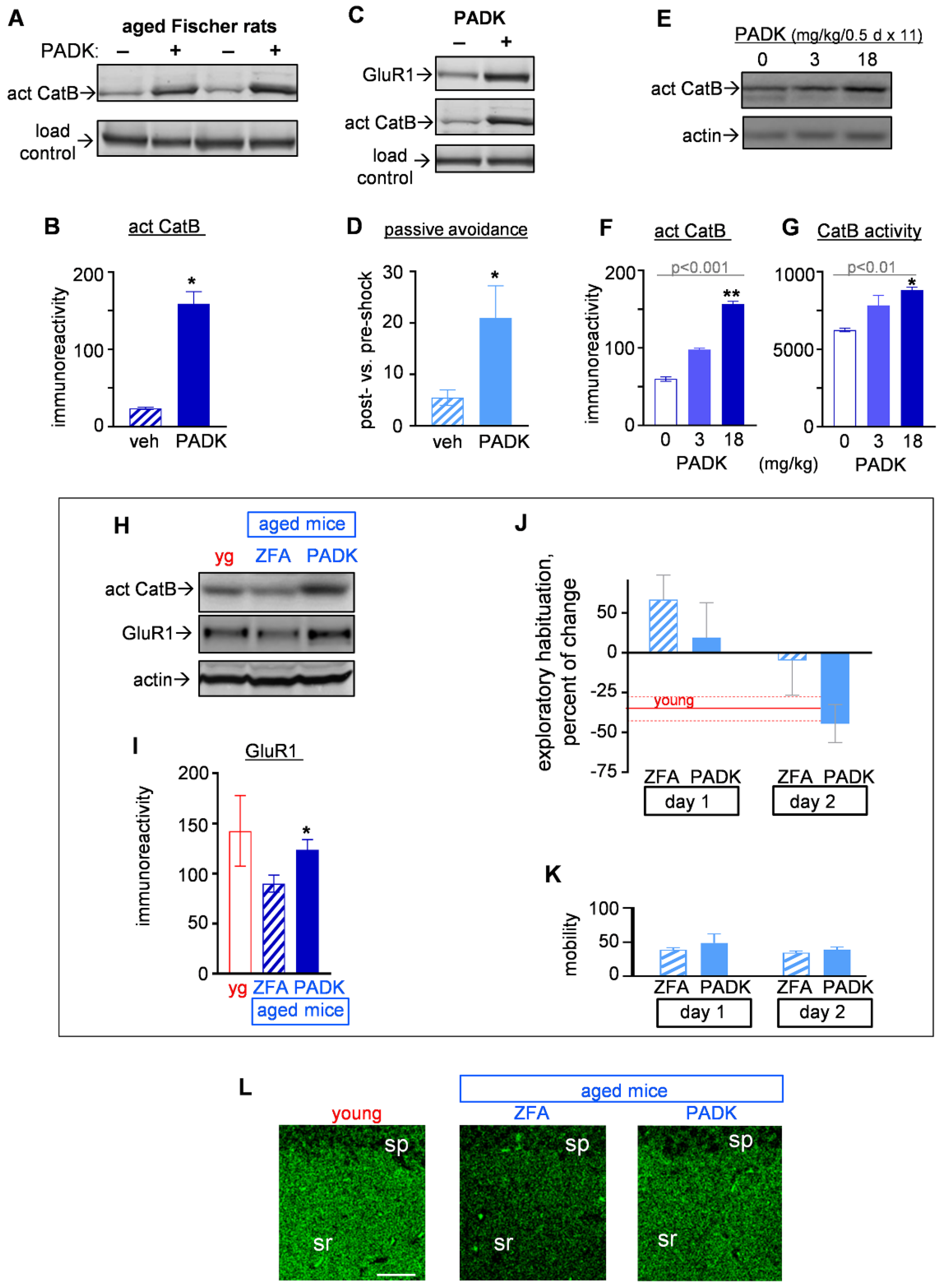
2.3.1. CatB Enhancement by PADK for Synaptic and Functional Improvements in Aged Female Fischer Rats
2.3.2. CatB Enhancement by PADK for Synaptic and Functional Improvements in Aged Mice
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Treatments
4.3. Behavioral Analyses
4.4. Sample Preparation for Analyses
4.5. Immunoblot Analysis
4.6. ELISA
4.7. Immunohistochemistry
4.8. Cathepsin B Activity and Proteasome Activity
4.9. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| act Cat B | 30-kDa active cathepsin B |
| Actb | β-actin gene |
| AD | Alzheimer’s disease |
| APP | amyloid precursor protein |
| Cat B | cathepsin B |
| CTF | carboxyl terminal fragment |
| CPP | conditioned place preference |
| DG | dentate gyrus |
| H&E | hematoxylin and eosin |
| iPSC | induced pluripotent stem cell |
| LAMP1 | lysosomal-associated membrane protein 1 |
| MCI | mild cognitive impairment |
| mf | mossy fibers |
| ml | molecular layer |
| NCAM | neural cell adhesion molecule |
| PADK | Z-Phe-Ala-diazomethylketone |
| PCR | polymerase chain reaction |
| PS1 | presenilin 1 |
| qRT-PCR | real-time quantitative reverse transcription PCR |
| SAB | spontaneous alternation behavior |
| sg | stratum granulosum |
| so | stratum oriens |
| sp | stratum pyramidale |
| veh | vehicle |
| WT | wild type |
| yg | young |
| ZFA | Z-Phe-Ala-OH |
References
- Bahr, B.A.; Wisniewski, M.L.; Butler, D. Positive lysosomal modulation as a unique strategy to treat age-related protein accumulation diseases. Rejuvenation Res. 2012, 15, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 2016, 32, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Telpoukhovskaia, M.A.; Bahr, B.A.; Chen, X.; Gan, L. Endo-lysosomal dysfunction: A converging mechanism in neurodegenerative diseases. Curr. Opin. Neurobiol. 2018, 48, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.; Bahr, B.A. Oxidative stress and lysosomes: CNS-related consequences and implications for lysosomal enhancement strategies and induction of autophagy. Antioxid. Redox Signal. 2006, 8, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Champ, C.; Day, J.; Aarts, E.; Bahr, B.A.; Bakker, M.; Banati, D.; Calabrese, V.; Cederholm, T.; Cryan, J.; et al. Poor cognitive ageing: Vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res. Rev. 2018, 42, 40–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Minakaki, G.; Nguyen, M.; Krainc, D. Preserving lysosomal function in the aging brain: Insights from neurodegeneration. Neurotherapeutics 2019. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef]
- Cecarini, V.; Bonfili, L.; Cuccioloni, M.; Mozzicafreddo, M.; Rossi, G.; Buizza, L.; Uberti, D.; Angeletti, M.; Eleuteri, A.M. Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Farizatto, K.L.G.; Ikonne, U.S.; Almeida, M.F.; Ferrari, M.F.R.; Bahr, B.A. Aβ42-mediated proteasome inhibition and associated tau pathology in hippocampus are governed by a lysosomal response involving cathepsin B: Evidence for protective crosstalk between protein clearance pathways. PLoS ONE 2017, 12, e0182895. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: Implications for Alzheimer’s disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.; Hwang, J.; Estick, C.; Nishiyama, A.; Kumar, S.S.; Baveghems, C.; Young-Oxendine, H.B.; Wisniewski, M.L.; Charalambides, A.; Bahr, B.A. Protective effects of positive lysosomal modulation in Alzheimer’s disease transgenic mouse models. PLoS ONE 2011, 6, e20501. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sun, B.; Zhou, Y.; Grubb, A.; Gan, L. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 2012, 287, 39834–39841. [Google Scholar] [CrossRef]
- Bendiske, J.; Bahr, B.A. Lysosomal activation is a compensatory response against protein accumulation and associated synaptopathogenesis-an approach for slowing Alzheimer disease? J. Neuropathol. Exp. Neurol. 2003, 62, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Qi, L.; Wang, Y.; Kegel, K.B.; Yoder, J.; Difiglia, M.; Qin, Z.H.; Lin, F. The regulation of N-terminal Huntingtin (Htt552) accumulation by Beclin1. Acta Pharm. Sin. 2012, 33, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Cecarini, V.; Bonfili, L.; Cuccioloni, M.; Mozzicafreddo, M.; Rossi, G.; Keller, J.N.; Angeletti, M.; Eleuteri, A.M. Wild type and mutant amyloid precursor proteins influence downstream effects of proteasome and autophagy inhibition. Biochim. Biophys. Acta 2014, 1842, 127–134. [Google Scholar] [CrossRef]
- Gavilán, E.; Pintado, C.; Gavilan, M.P.; Daza, P.; Sanchez-Aguayo, I.; Castano, A.; Ruano, D. Age-related dysfunctions of the autophagy lysosomal pathway in hippocampal pyramidal neurons under proteasome stress. Neurobiol. Aging 2015, 36, 1953–1963. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Hamilton, D.J.; Barnett, J.L.; Paskevich, P.A.; Nixon, R.A. Properties of the endosomal-lysosomal system in the human central nervous system: Disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J. Neurosci. 1996, 16, 186–199. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Sundelöf, J.; Sundström, J.; Hansson, O.; Eriksdotter-Jönhagen, M.; Giedraitis, V.; Larsson, A.; Degerman-Gunnarsson, M.; Ingelsson, M.; Minthon, L.; Blennow, K.; et al. Higher cathepsin B levels in plasma in Alzheimer’s disease compared to healthy controls. J. Alzheimers Dis. 2010, 22, 1223–1230. [Google Scholar] [CrossRef]
- Morena, F.; Argentati, C.; Trotta, R.; Crispoltoni, L.; Stabile, A.; Pistilli, A.; di Baldassarre, A.; Calafiore, R.; Montanucci, P.; Basta, G.; et al. A comparison of lysosomal enzymes expression levels in peripheral blood of mild- and severe-Alzheimer’s disease and MCI patients: Implications for regenerative medicine approaches. Int. J. Mol. Sci. 2017, 18, 1806. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.Y.; Becke, A.; Berron, D.; Becker, B.; Sah, N.; Benoni, G.; Janke, E.; Lubejko, S.T.; Greig, N.H.; Mattison, J.A.; et al. Running-induced systemic cathepsin B secretion is associated with memory function. Cell Metab. 2016, 24, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Trinchese, F.; Fá, M.; Liu, S.; Zhang, H.; Hidalgo, A.; Schmidt, S.D.; Yamaguchi, H.; Yoshii, N.; Mathews, P.M.; Nixon, R.A.; et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J. Clin. Invest. 2008, 118, 2796–2807. [Google Scholar] [CrossRef] [PubMed]
- Romine, H.; Rentschler, K.M.; Smith, K.; Edwards, A.; Colvin, C.; Farizatto, K.; Pait, M.C.; Butler, D.; Bahr, B.A. Potential Alzheimer’s disease therapeutics among weak cysteine protease inhibitors exhibit mechanistic differences regarding extent of cathepsin B up-regulation and ability to block calpain. Eur. Sci. J. 2017, 13, 38–59. [Google Scholar] [CrossRef]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to β-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef]
- Saito, T.; Matsuba, Y.; Mihira, N.; Takano, J.; Nilsson, P.; Itohara, S.; Iwata, N.; Saido, T.C. Single APP knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 2014, 17, 661–663. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Robbins, E.M.; Zhang-Nunes, S.X.; Purcell, S.M.; Betensky, R.A.; Raju, S.; Prada, C.; Greenberg, S.M.; Bacskai, B.J.; Frosch, M.P. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 2006, 24, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Barrachina, M.; Maes, T.; Buesa, C.; Ferrer, I. Lysosome-associated membrane protein 1 (LAMP-1) in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2006, 32, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Yuan, P.; Wu, Y.; Schrag, M.; Paradise, S.; Grutzendler, J.; De Camilli, P.; Ferguson, S.M. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. USA 2015, 112, E3699–E3708. [Google Scholar] [CrossRef] [PubMed]
- Mackay, E.A.; Ehrhard, A.; Moniatte, M.; Guenet, C.; Tardif, C.; Tarnus, C.; Sorokine, O.; Heintzelmann, B.; Nay, C.; Remy, J.M.; et al. A possible role for cathepsins D, E, and B in the processing of β-amyloid precursor protein in Alzheimer’s disease. Eur. J. Biochem. 1997, 244, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, I.; Pardossi-Piquard, R.; Bourgeois, A.; Pagnotta, S.; Biferi, M.G.; Barkats, M.; Lacor, P.; Klein, W.; Bauer, C.; Checler, F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta. Neuropathol. 2016, 132, 257–276. [Google Scholar] [CrossRef]
- Palop, J.J.; Jones, B.; Kekonius, L.; Chin, J.; Yu, G.Q.; Raber, J.; Masliah, E.; Mucke, L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc. Natl. Acad. Sci. USA 2003, 100, 9572–9577. [Google Scholar] [CrossRef] [PubMed]
- Kojetin, D.J.; Venters, R.A.; Kordys, D.R.; Thompson, R.J.; Kumar, R.; Cavanagh, J. Structure, binding interface and hydrophobic transitions of Ca2+-loaded calbindin-D(28K). Nat. Struct. Mol. Biol. 2006, 13, 641–647. [Google Scholar] [CrossRef]
- Schmidt, H. Three functional facets of calbindin D-28k. Front. Mol. Neurosci. 2012, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Pepeu, G. Mild cognitive impairment: Animal models. Dialogues Clin. Neurosci. 2004, 6, 369–377. [Google Scholar]
- Bizon, J.L.; LaSarge, C.L.; Montgomery, K.S.; McDermott, A.N.; Setlow, B.; Griffith, W.H. Spatial reference and working memory across the lifespan of male Fischer 344 rats. Neurobiol. Aging 2009, 30, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Counts, S.E.; He, B.; Nadeem, M.; Wuu, J.; Scheff, S.W.; Mufson, E.J. Hippocampal drebrin loss in mild cognitive impairment. Neurodegener Dis. 2012, 10, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Neuner, S.M.; Ding, S.; Kaczorowski, C.C. Knockdown of heterochromatin protein 1 binding protein 3 recapitulates phenotypic, cellular, and molecular features of aging. Aging Cell 2019, 18, e12886. [Google Scholar] [CrossRef] [PubMed]
- Bahr, B.A.; Bendiske, J. The neuropathogenic contributions of lysosomal dysfunction. J. Neurochem. 2002, 83, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Schain, A.; Grutzendler, J. Multicolor time-stamp reveals the dynamics and toxicity of amyloid deposition. Sci. Rep. 2011, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, A.M.; Nixon, R.A. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc. Natl. Acad. Sci. USA 1990, 87, 3861–3865. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, S.D.; Alldred, M.J.; Counts, S.E.; Cataldo, A.M.; Neve, R.L.; Jiang, Y.; Wuu, J.; Chao, M.V.; Mufson, E.J.; Nixon, R.A.; et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 2010, 68, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Bahr, B.A.; Hoffman, K.B.; Yang, A.J.; Hess, U.S.; Glabe, C.G.; Lynch, G. Amyloid β protein is internalized selectively by hippocampal field CA1 and causes neurons to accumulate amyloidogenic carboxyterminal fragments of the amyloid precursor protein. J. Comp. Neurol. 1998, 397, 139–147. [Google Scholar] [CrossRef]
- Wisniewski, M.L.; Hwang, J.; Bahr, B.A. Submicromolar Aβ42 reduces hippocampal glutamate receptors and presynaptic markers in an aggregation-dependent manner. Biochim. Biophys. Acta 2011, 1812, 1664–1674. [Google Scholar] [CrossRef]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal dysfunction in Down syndrome is APP-dependent and mediated by APP-βCTF (C99). J. Neurosci. 2019, 39, 5255–5268. [Google Scholar] [CrossRef]
- Perez, S.E.; He, B.; Nadeem, M.; Wuu, J.; Ginsberg, S.D.; Ikonomovic, M.D.; Mufson, E.J. Hippocampal endosomal, lysosomal, and autophagic dysregulation in mild cognitive impairment: Correlation with Aβ and tau pathology. J. Neuropathol. Exp. Neurol. 2015, 74, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Bendiske, J.; Caba, E.; Brown, Q.B.; Bahr, B.A. Intracellular deposition, microtubule destabilization, and transport failure: An “early” pathogenic cascade leading to synaptic decline. J. Neuropathol. Exp. Neurol. 2002, 61, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.; Brown, Q.B.; Chin, D.J.; Batey, L.; Karim, S.; Mutneja, M.S.; Karanian, D.A.; Bahr, B.A. Cellular responses to protein accumulation involve autophagy and lysosomal enzyme activation. Rejuvenation Res. 2005, 8, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Scheff, S.W.; Styren, S.D. Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration 1996, 5, 417–421. [Google Scholar] [CrossRef]
- Coleman, P.D.; Yao, P.J. Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 1023–1027. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Zheng, X.; Gessel, M.M.; Wisniewski, M.L.; Viswanathan, K.; Wright, D.L.; Bahr, B.A.; Bowers, M.T. Z-Phe-Ala-diazomethylketone (PADK) disrupts and remodels early oligomer states of the Alzheimer disease Aβ42 protein. J. Biol. Chem. 2012, 287, 6084–6088. [Google Scholar] [CrossRef]
- Boland, B.; Yu, W.H.; Corti, O.; Mollereau, B.; Henriques, A.; Bezard, E.; Pastores, G.M.; Rubinsztein, D.C.; Nixon, R.A.; Duchen, M.R.; et al. Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug. Discov. 2018, 17, 660–688. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy activator drugs: A new opportunity in neuroprotection from misfolded protein toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Weissmiller, A.M.; White, J.A.; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Invest. 2016, 126, 1815–1833. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, K.; Hoover, D.J.; Hwang, J.; Wisniewski, M.L.; Ikonne, U.S.; Bahr, B.A.; Wright, D.L. Nonpeptidic lysosomal modulators derived from Z-Phe-Ala-diazomethylketone for treating protein accumulation diseases. ACS Med. Chem. Lett. 2012, 3, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Embury, C.M.; Dyavarshetty, B.; Lu, Y.; Wiederin, J.L.; Ciborowski, P.; Gendelman, H.E.; Kiyota, T. Cathepsin B improves β-amyloidosis and learning and memory in models of Alzheimer’s disease. J. Neuroimmune Pharm. Pharmacol. 2017, 12, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Ryzhikov, S.; Bahr, B.A. Gephyrin alterations due to protein accumulation stress are reduced by the lysosomal modulator Z-Phe-Ala-diazomethylketone. J. Mol. Neurosci. 2008, 34, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Jana, M.; Pahan, K. Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of Alzheimer’s disease via PPARα. J. Neurosci. 2018, 38, 6682–6699. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Machhi, J.; Lu, Y.; Dyavarshetty, B.; Nemati, M.; Zhang, G.; Mosley, R.L.; Gelbard, H.A.; Gendelman, H.E. URMC-099 facilitates amyloid-β clearance in a murine model of Alzheimer’s disease. J. Neuroinflammation 2018, 15, 137. [Google Scholar] [CrossRef]
- Boutry, M.; Branchu, J.; Lustremant, C.; Pujol, C.; Pernelle, J.; Matusiak, R.; Seyer, A.; Poirel, M.; Chu-Van, E.; Pierga, A.; et al. Inhibition of lysosome membrane recycling causes accumulation of gangliosides that contribute to neurodegeneration. Cell Rep. 2018, 23, 3813–3826. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gelé, P.; Drobeck, H.; Bégard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C.; Buée, L.; et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef]
- Eitan, E.; Suire, C.; Zhang, S.; Mattson, M.P. Impact of lysosome status on extracellular vesicle content and release. Ageing Res. Rev. 2016, 32, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Tancini, B.; Buratta, S.; Sagini, K.; Costanzi, E.; Delo, F.; Urbanelli, L.; Emiliani, C. Insight into the role of extracellular vesicles in lysosomal storage disorders. Genes (Basel) 2019, 10, 510. [Google Scholar] [CrossRef] [PubMed]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wong, Y.C.; Ysselstein, D.; Severino, A.; Krainc, D. Synaptic, mitochondrial, and lysosomal dysfunction in Parkinson’s disease. Trends Neurosci. 2019, 42, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bové, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. α-Synuclein (αSyn) preformed fibrils induce endogenous αSyn aggregation, compromise synaptic activity and enhance synapse loss in cultured excitatory hippocampal neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef]
- Batey, L.; Chin, D.J.; Karim, S.; Karanian, D.A.; Butler, D.; Bahr, B.A. Lysosomal modulation and autophagic induction: Compensatory processes in response to protein deposition pathology. In In New Trends in Alzheimer and Parkinson Related Disorders: ADPD 2005; Medimond International Proceedings: Bologna, Italy, 2005; pp. 55–58. [Google Scholar]
- Bahr, B.A. Lysosomal modulation: A way to treat brain aging and age-related disorders? In In Aging of Brain and Alzheimer Disease: Brain Aging Science; Fundan Press: Shanghai, China, 2008; Vol. 2, pp. 331–333. [Google Scholar]
- Bahr, B.A. Lysosomal modulatory drugs for a broad strategy against protein accumulation disorders. Curr. Alzheimer Res. 2009, 6, 438–445. [Google Scholar] [CrossRef]
- Tsunemi, T.; Perez-Rosello, T.; Ishiguro, Y.; Yoroisaka, A.; Jeon, S.; Hamada, K.; Wong, Y.C.; Xie, Z.; Akamatsu, W.; Mazzulli, J.R.; et al. Increased lysosomal exocytosis induced by lysosomal Ca(2+) channel agonists protects human dopaminergic neurons from α-synuclein toxicity. J. Neurosci. 2019. [Google Scholar] [CrossRef]
- Petersen, R.C.; Roberts, R.O.; Knopman, D.S.; Boeve, B.F.; Geda, Y.E.; Ivnik, R.J.; Smith, G.E.; Jack, C.R., Jr. Mild cognitive impairment: Ten years later. Arch. Neurol. 2009, 66, 1447–1455. [Google Scholar] [CrossRef]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: Role of Aβ in pathogenesis. Acta Neuropathol. 2009, 118, 131–150. [Google Scholar] [CrossRef]
- Jang, Y.; Kwon, I.; Song, W.; Cosio-Lima, L.M.; Lee, Y. Endurance exercise mediates neuroprotection against MPTP-mediated Parkinson’s disease via enhanced neurogenesis, antioxidant capacity, and autophagy. Neuroscience 2018, 379, 292–301. [Google Scholar] [CrossRef]
- Takahashi, H.; Yamazaki, H.; Hanamura, K.; Sekino, Y.; Shirao, T. Activity of the AMPA receptor regulates drebrin stabilization in dendritic spine morphogenesis. J. Cell Sci. 2009, 122, 1211–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, C.; Zaleska, M.M.; Riddell, D.R.; Atchison, K.P.; Robshaw, A.; Zhou, H.; Sukoff Rizzo, S.J. Alternative method of oral administration by peanut butter pellet formulation results in target engagement of BACE1 and attenuation of gavage-induced stress responses in mice. Pharm. Biochem. Behav. 2014, 126, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Kosten, T.A.; Kim, J.J.; Lee, H.J. Early life manipulations alter learning and memory in rats. Neurosci. Biobehav. Rev. 2012, 36, 1985–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.; Piehler, T.; Benjamin, R.; Farizatto, K.L.; Pait, M.C.; Almeida, M.F.; Ghukasyan, V.V.; Bahr, B.A. Blast waves from detonated military explosive reduce GluR1 and synaptophysin levels in hippocampal slice cultures. Exp. Neurol. 2016, 286, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farizatto, K.L.G.; Almeida, M.F.; Long, R.T.; Bahr, B.A. Early synaptic alterations and selective adhesion signaling in hippocampal dendritic zones following organophosphate exposure. Sci. Rep. 2019, 9, 6532. [Google Scholar] [CrossRef] [PubMed]
- Bahr, B.A.; Hoffman, K.B.; Kessler, M.; Hennegriff, M.; Park, G.Y.; Yamamoto, R.S.; Kawasaki, B.T.; Vanderklish, P.W.; Hall, R.A.; Lynch, G. Distinct distributions of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor subunits and a related 53,000 M(R) antigen (GR53) in brain tissue. Neuroscience 1996, 74, 707–721. [Google Scholar] [CrossRef]
- Royer-Zemmour, B.; Ponsole-Lenfant, M.; Gara, H.; Roll, P.; Lévêque, C.; Massacrier, A.; Ferracci, G.; Cillario, J.; Robaglia-Schlupp, A.; Vincentelli, R.; et al. Epileptic and developmental disorders of the speech cortex: Ligand/receptor interaction of wild-type and mutant SRPX2 with the plasminogen activator receptor uPAR. Hum. Mol. Genet. 2008, 17, 3617–3630. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Weight (g/mol) | CatB Change | GluR1 Recovery |
|---|---|---|---|
| PADK | 394.4 | 4.9-fold increase | 100% |
| SD1002 | 446.5 | 3.1-fold increase | 100% |
| BW1005 | 496.5 | 2.8-fold increase | 100% |
| UP1A101 | 328.4 | 2.1-fold increase | 53% |
| E64d | 342.4 | 1.7-fold increase | 39% |
| Measurements | Results Determined after PADK Treatment |
|---|---|
| LAMP1-positive organelles/neuron | 97 ± 9% of vehicle-treated mice (N.S.) |
| CatB-positive organelles/neuron | 98 ± 5% of vehicle-treated mice (N.S.) |
| CatB mRNA in transgenic mice | −0.497 ΔΔCt compared to vehicle-treated mice (N.S.) |
| CatB mRNA in Fischer rats | −0.599 ΔΔCt compared to vehicle-treated rats (N.S.) |
| Proteasome activity | 117 ± 12% of vehicle-treated controls (N.S.) |
| SRPX2 CatB-interacting protein | 95 ± 6% of vehicle-treated controls (N.S.) |
| Body weight of 9-month female mice | 103.7 ± 1.9% of start weight (N.S.) |
| Body weight of 9-month male mice | 104.9 ± 1.4% of start weight (N.S.) |
| Body weight of 22-month male mice | 101.8 ± 0.6% of start weight (N.S.) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, J.; Estick, C.M.; Ikonne, U.S.; Butler, D.; Pait, M.C.; Elliott, L.H.; Ruiz, S.; Smith, K.; Rentschler, K.M.; Mundell, C.; et al. The Role of Lysosomes in a Broad Disease-Modifying Approach Evaluated across Transgenic Mouse Models of Alzheimer’s Disease and Parkinson’s Disease and Models of Mild Cognitive Impairment. Int. J. Mol. Sci. 2019, 20, 4432. https://doi.org/10.3390/ijms20184432
Hwang J, Estick CM, Ikonne US, Butler D, Pait MC, Elliott LH, Ruiz S, Smith K, Rentschler KM, Mundell C, et al. The Role of Lysosomes in a Broad Disease-Modifying Approach Evaluated across Transgenic Mouse Models of Alzheimer’s Disease and Parkinson’s Disease and Models of Mild Cognitive Impairment. International Journal of Molecular Sciences. 2019; 20(18):4432. https://doi.org/10.3390/ijms20184432
Chicago/Turabian StyleHwang, Jeannie, Candice M. Estick, Uzoma S. Ikonne, David Butler, Morgan C. Pait, Lyndsie H. Elliott, Sarah Ruiz, Kaitlan Smith, Katherine M. Rentschler, Cary Mundell, and et al. 2019. "The Role of Lysosomes in a Broad Disease-Modifying Approach Evaluated across Transgenic Mouse Models of Alzheimer’s Disease and Parkinson’s Disease and Models of Mild Cognitive Impairment" International Journal of Molecular Sciences 20, no. 18: 4432. https://doi.org/10.3390/ijms20184432