Differential Expression Profiles of Oxidative Stress Levels, 8-oxo-dG and 4-HNE, in Barrett’s Esophagus Compared to Esophageal Adenocarcinoma

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Differences in Oxidative Stress across the BE-Disease Sequence
2.1.1. Aged-Matched Histology Groups
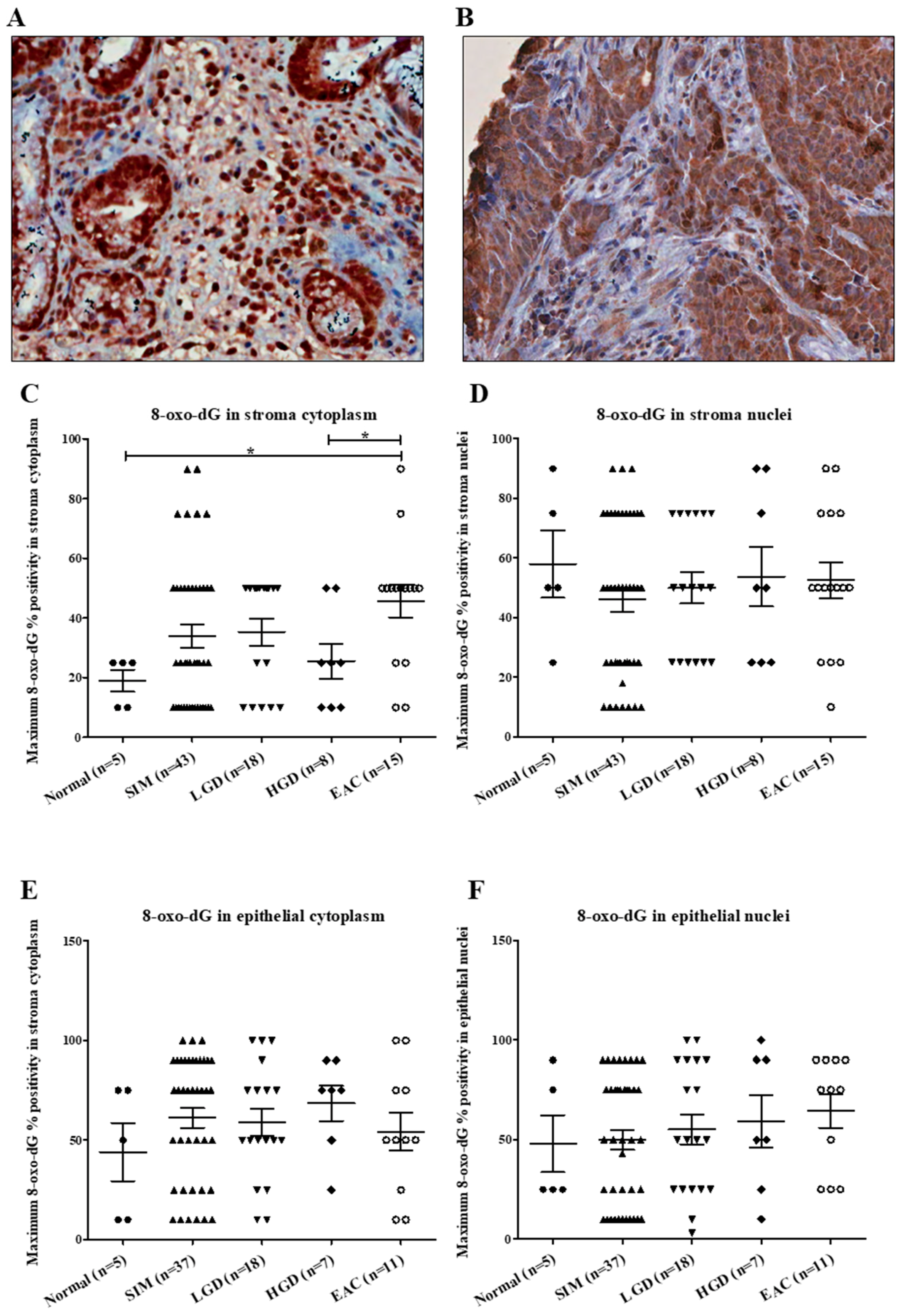
2.1.2. 8-oxo-dG Expression along the BE Disease Sequence
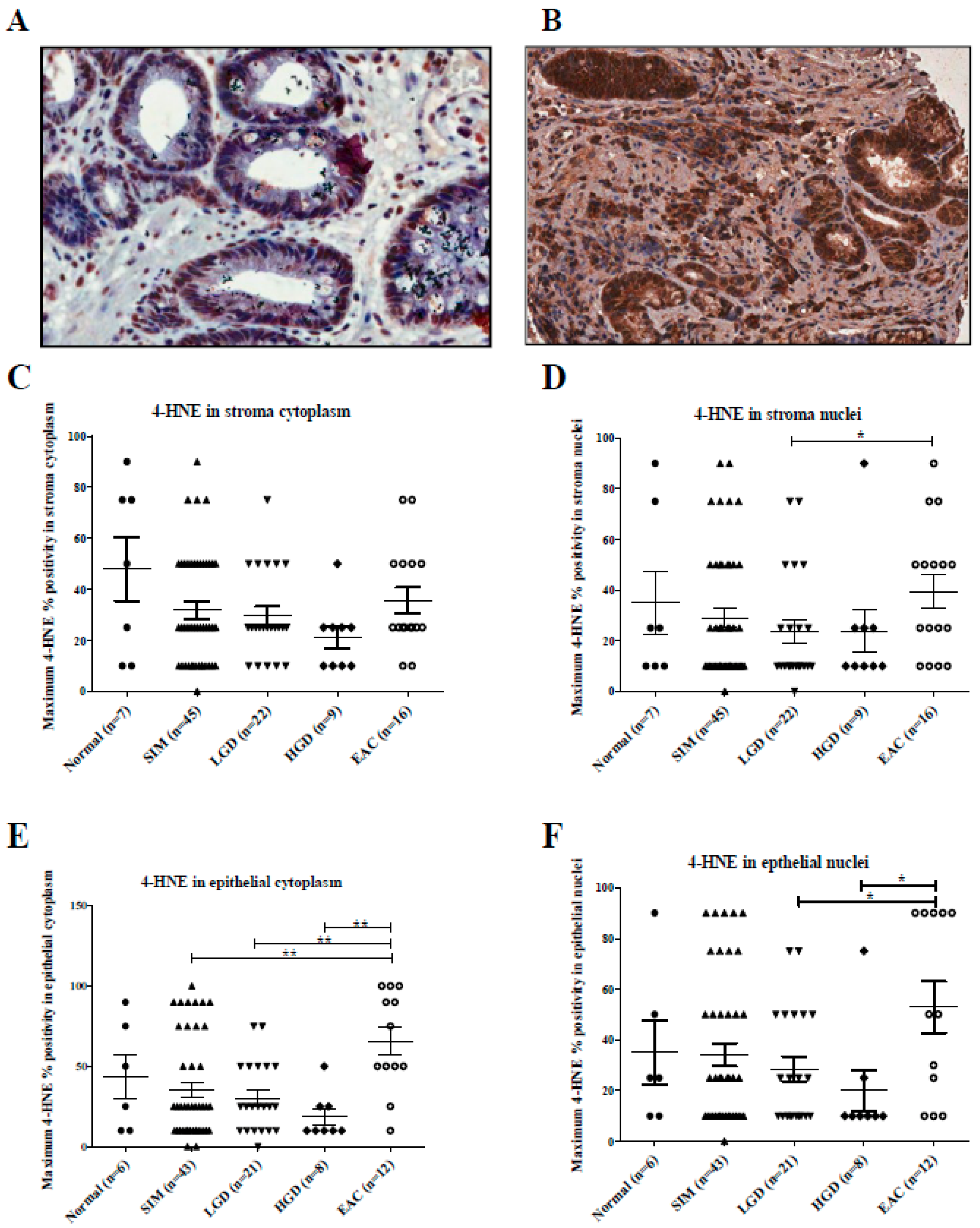
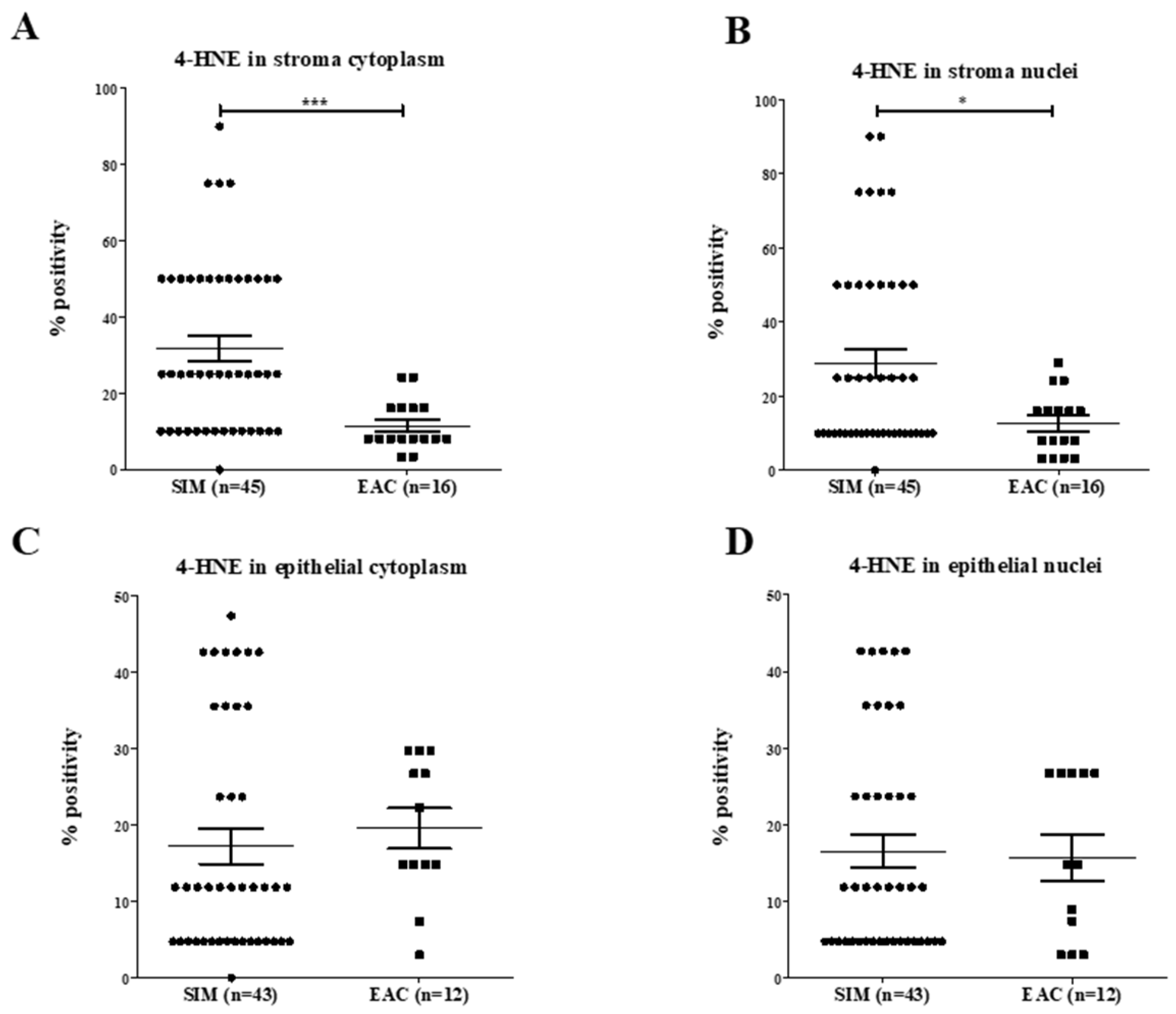
2.1.3. 4-HNE Expression Along the BE Disease Sequence
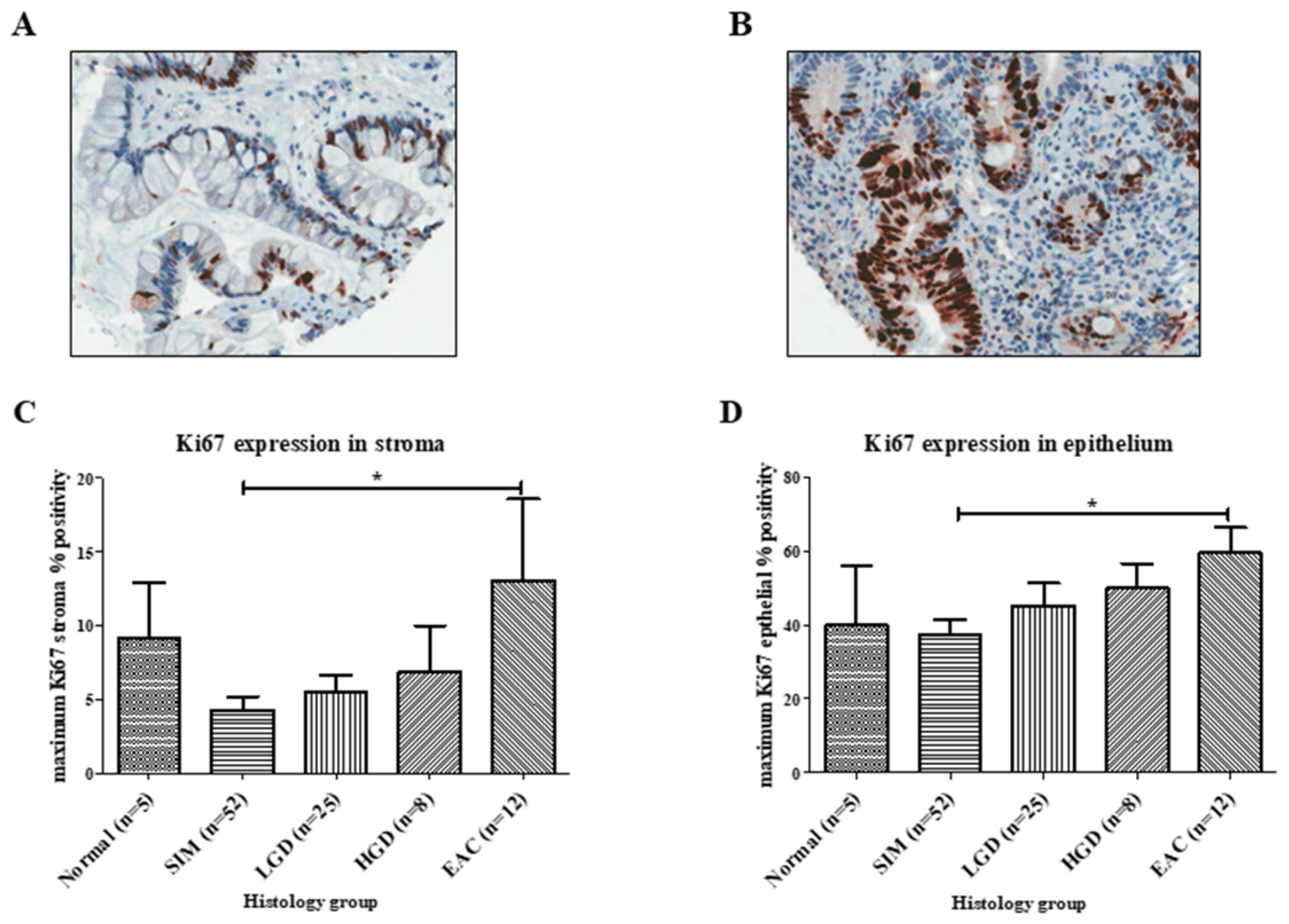
2.1.4. Ki67 Expression along the BE Disease Sequence
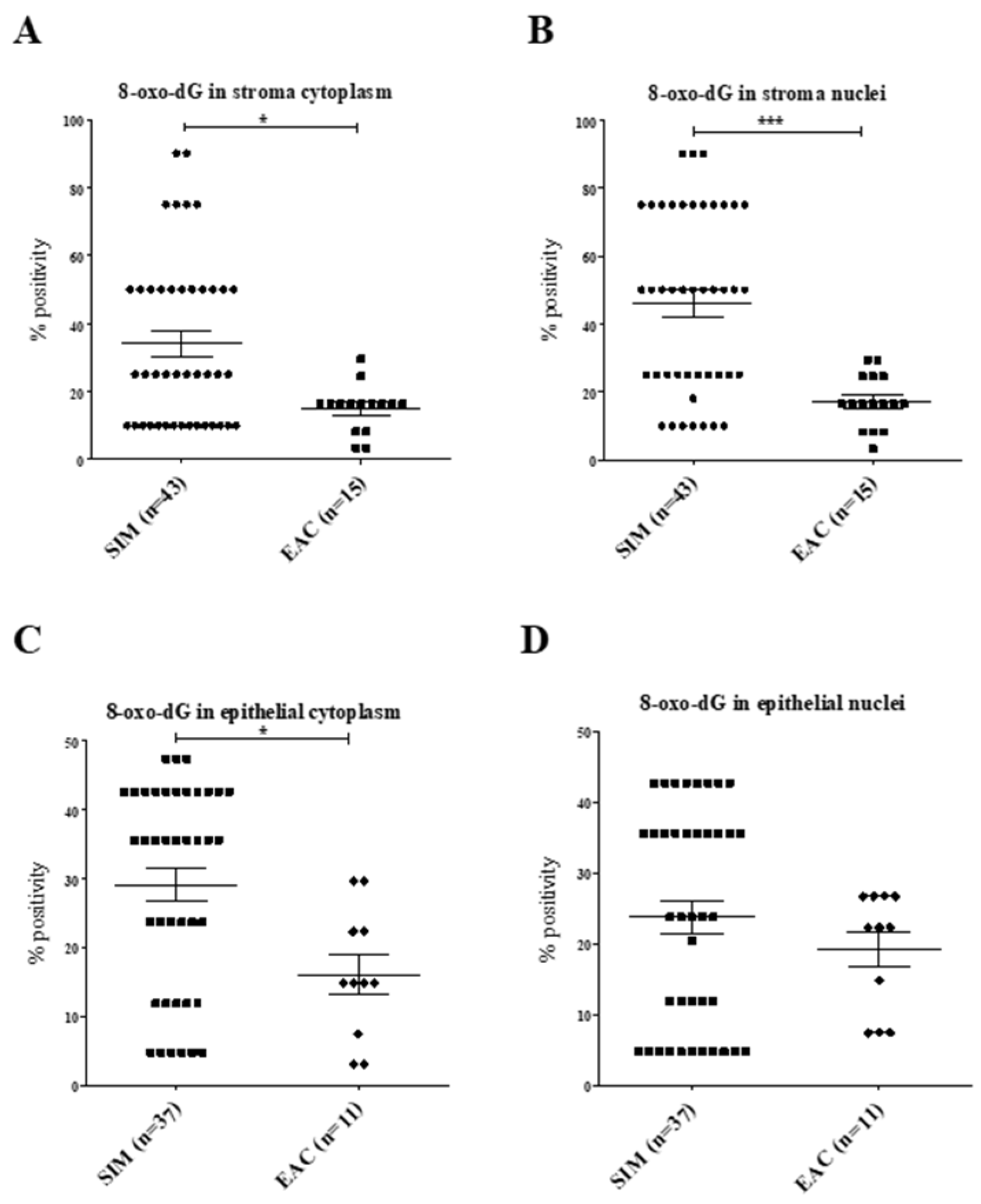
2.1.5. Normalization of 8-oxo-dG Expression to Proliferation in SIM versus EAC
2.1.6. Normalization of 4-HNE Expression to Proliferation in SIM versus EAC
2.2. Differences in Proliferation (Ki67), Oxidative Stress (8-oxo-dG) and Lipoperoxidation (4-HNE) between SIM Progressors and non-Progressors
2.2.1. Demographics of SIM Progressors and Non-Progressors
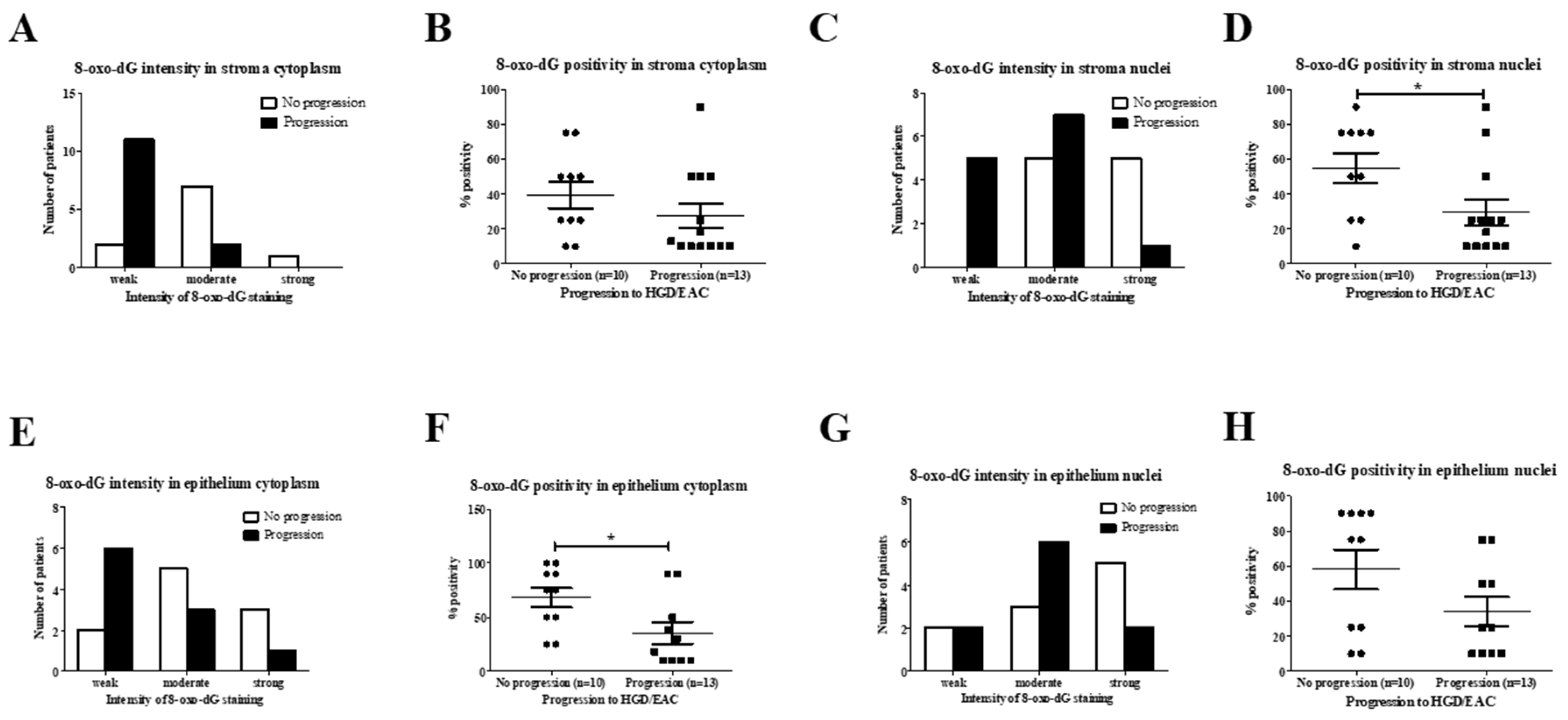
2.2.2. Analysis of 8-oxo-dG in SIM Progressors versus Non-Progressors
2.2.3. Analysis of 4-HNE in SIM Progressors versus Non-Progressors
2.2.4. Ki67 Expression in SIM Progressors versus Non-Progressors
3. Discussion
4. Materials and Methods
4.1. Patient Samples
4.2. TMA Construction
4.3. 8-oxo-dG and 4-HNE Immunohistochemistry
4.4. Ki67 Immunohistochemistry
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Barrett’s esophagus | BE |
| Esophageal adenocarcinoma | EAC |
| specialized intestinal metaplasia of Barrett’s esophagus | SIM |
| low grade dysplasia | LGD |
| high grade dysplasia | HGD |
| reactive oxygen species | ROS |
| deoxyribonucleic acid | DNA |
| deoxycholic acid | DCA |
| 8-oxo-7, 8-dihydro-2’-deoxyguanine | 8-oxo-dG |
| 4-hydroxy-2-nonenal | 4-HNE |
| hydroxyl | OH |
| nuclear factor-kappa beta | NF-κB |
| tissue microarray | TMA |
| Haematoxylin- and eosin | H&E |
| hydrogen peroxide | H2O2 |
| phosphate buffered saline | PBS |
| diaminobenzidine | DAB |
| dibutylphthalate polystyrene xylene | DPX |
References
- Blot, W.J.; Devesa, S.S.; Kneller, R.W.; Fraumeni, J.F., Jr. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. J. Am. Med Assoc. 1991, 265, 1287–1289. [Google Scholar] [CrossRef]
- Jankowski, J.A.; Perry, I.; Harrison, R.F. Gastro-oesophageal cancer: Death at the junction. Br. Med. J. 2000, 321, 463–464. [Google Scholar] [CrossRef]
- Van Soest, E.M.; Dieleman, J.P.; Siersema, P.D.; Sturkenboom, M.C.; Kuipers, E.J. Increasing incidence of barrett’s oesophagus in the general population. Gut 2005, 54, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.J.; Ott, B.J.; Payne, W.S. The incidence of adenocarcinoma in columnar-lined (barrett’s) esophagus. N. Engl. J. Med. 1985, 313, 857–859. [Google Scholar] [CrossRef] [PubMed]
- Spechler, S.J.; Goyal, R.K. Barrett’s esophagus. N. Engl. J. Med. 1986, 315, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.H.; Clark, G.W.; Ireland, A.P.; Chandrasoma, P.; Smyrk, T.C.; DeMeester, T.R. Outcome of adenocarcinoma arising in barrett’s esophagus in endoscopically surveyed and nonsurveyed patients. J. Thorac. Cardiovasc. Surg. 1994, 108, 813–821. [Google Scholar] [PubMed]
- Van Sandick, J.W.; van Lanschot, J.J.; Kuiken, B.W.; Tytgat, G.N.; Offerhaus, G.J.; Obertop, H. Impact of endoscopic biopsy surveillance of barrett’s oesophagus on pathological stage and clinical outcome of barrett’s carcinoma. Gut 1998, 43, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sorensen, H.T.; Funch-Jensen, P. Incidence of adenocarcinoma among patients with barrett’s esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.M.; Rowley, S.P.; Fitzgerald, A.P.; Ravi, N.; Reynolds, J.V. Adenocarcinoma of the oesophagus and gastric cardia: Male preponderance in association with obesity. Eur. J. Cancer 2006, 42, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.B.; Falk, G.W.; Richter, J.E. The incidence of adenocarcinoma and dysplasia in barrett’s esophagus: Report on the cleveland clinic barrett’s esophagus registry. Am. J. Gastroenterol. 1999, 94, 2037–2042. [Google Scholar] [PubMed]
- Pepe, M.S.; Etzioni, R.; Feng, Z.; Potter, J.D.; Thompson, M.L.; Thornquist, M.; Winget, M.; Yasui, Y. Phases of biomarker development for early detection of cancer. J. Natl. Cancer Inst. 2001, 93, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Timmer, M.R.; Sun, G.; Gorospe, E.C.; Leggett, C.L.; Lutzke, L.; Krishnadath, K.K.; Wang, K.K. Predictive biomarkers for barrett’s esophagus: So near and yet so far. Dis. Esophagus 2013, 26, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Bird-Lieberman, E.L.; Dunn, J.M.; Coleman, H.G.; Lao-Sirieix, P.; Oukrif, D.; Moore, C.E.; Varghese, S.; Johnston, B.T.; Arthur, K.; McManus, D.T.; et al. Population-based study reveals new risk-stratification biomarker panel for barrett’s esophagus. Gastroenterology 2012, 143, 927–935. [Google Scholar] [CrossRef]
- Maley, C.C.; Galipeau, P.C.; Finley, J.C.; Wongsurawat, V.J.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Blount, P.L.; Risques, R.A.; Rabinovitch, P.S.; et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat. Genet. 2006, 38, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Merlo, L.M.; Shah, N.A.; Li, X.; Blount, P.L.; Vaughan, T.L.; Reid, B.J.; Maley, C.C. A comprehensive survey of clonal diversity measures in barrett’s esophagus as biomarkers of progression to esophageal adenocarcinoma. Cancer Prev. 2010, 3, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G. Mitochondria in cancer. Oncogene 2006, 25, 4630–4632. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A. Mitochondrial DNA and ageing. Biochim. Biophys. Acta 2006, 1757, 611–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; Van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef]
- Trifunovic, A.; Hansson, A.; Wredenberg, A.; Rovio, A.T.; Dufour, E.; Khvorostov, I.; Spelbrink, J.N.; Wibom, R.; Jacobs, H.T.; Larsson, N.G. Somatic mtdna mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. USA 2005, 102, 17993–17998. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 1992, 256, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Diseases of the mitochondrial DNA. Annu. Rev. Biochem. 1992, 61, 1175–1212. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, N.J.; Feighery, R.; Picardo, S.L.; Lynam-Lennon, N.; Biniecka, M.; McGarrigle, S.A.; Phelan, J.J.; MacCarthy, F.; O’Toole, D.; Fox, E.J.; et al. Changes in mitochondrial stability during the progression of the barrett’s esophagus disease sequence. BMC Cancer 2016, 16, 497. [Google Scholar] [CrossRef]
- Loeb, L.A. Mutator phenotype in cancer: Origin and consequences. Semin. Cancer Biol. 2010, 20, 279–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, G.J.; D’Souza, F.R.; Suzen, S.H.; Eltahir, Z.S.; James, S.A.; Parry, J.M.; Griffiths, P.A.; Baxter, J.N. Deoxycholic acid at neutral and acid ph, is genotoxic to oesophageal cells through the induction of ros: The potential role of anti-oxidants in barrett’s oesophagus. Carcinogenesis 2007, 28, 136–142. [Google Scholar] [CrossRef]
- Jenkins, G.J.; Harries, K.; Doak, S.H.; Wilmes, A.; Griffiths, A.P.; Baxter, J.N.; Parry, J.M. The bile acid deoxycholic acid (dca) at neutral ph activates nf-kappab and induces il-8 expression in oesophageal cells in vitro. Carcinogenesis 2004, 25, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ding, Y.W.; Yang, G.; Bondoc, F.; Lee, M.J.; Yang, C.S. Oxidative damage in an esophageal adenocarcinoma model with rats. Carcinogenesis 2000, 21, 257–263. [Google Scholar] [CrossRef] [Green Version]
- Inayama, M.; Hashimoto, N.; Tokoro, T.; Shiozaki, H. Involvement of oxidative stress in experimentally induced reflux esophagitis and esophageal cancer. Hepato Gastroenterol. 2007, 54, 761–765. [Google Scholar]
- Lee, S.; Han, M.J.; Lee, K.S.; Back, S.C.; Hwang, D.; Kim, H.Y.; Shin, J.H.; Suh, S.P.; Ryang, D.W.; Kim, H.R.; et al. Frequent occurrence of mitochondrial DNA mutations in barrett’s metaplasia without the presence of dysplasia. PLoS ONE 2012, 7, e37571. [Google Scholar] [CrossRef] [PubMed]
- Biniecka, M.; Kennedy, A.; Fearon, U.; Ng, C.T.; Veale, D.J.; O’Sullivan, J.N. Oxidative damage in synovial tissue is associated with in vivo hypoxic status in the arthritic joint. Ann. Rheum. Dis. 2010, 69, 1172–1178. [Google Scholar] [CrossRef]
- Biniecka, M.; Fox, E.; Gao, W.; Ng, C.T.; Veale, D.J.; Fearon, U.; O’Sullivan, J. Hypoxia induces mitochondrial mutagenesis and dysfunction in inflammatory arthritis. Arthritis Rheum. 2011, 63, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Moriya, M. Single-stranded shuttle phagemid for mutagenesis studies in mammalian cells: 8-oxoguanine in DNA induces targeted g.C-->t.A transversions in simian kidney cells. Proc. Natl. Acad. Sci. USA 1993, 90, 1122–1126. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, K.; Payne, C.M.; Chavarria, M.; Ramsey, L.; Dvorakova, B.; Bernstein, H.; Holubec, H.; Sampliner, R.E.; Guy, N.; Condon, A.; et al. Bile acids in combination with low ph induce oxidative stress and oxidative DNA damage: Relevance to the pathogenesis of barrett’s oesophagus. Gut 2007, 56, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Temma-Asano, K.; Tskitishvili, E.; Kanagawa, T.; Tomimatsu, T.; Tsutsui, T.; Kimura, T.; Chang, Y.S.; Nakamura, T.; Nakai, Y.; Shimoya, K. Effects of 4-hydroxy-2-nonenal, a major lipid peroxidation-derived aldehyde, and n-acetylcysteine on the cyclooxygenase-2 expression in human uterine myometrium. Gynecol. Obstet. Investig. 2011, 72, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Ho, C.T.; Liong, E.C.; Nanji, A.A.; Leung, T.M.; Lau, T.Y.; Fung, M.L.; Tipoe, G.L. Epigallocatechin gallate attenuates fibrosis, oxidative stress, and inflammation in non-alcoholic fatty liver disease rat model through tgf/smad, pi3 k/akt/foxo1, and nf-kappa b pathways. Eur. J. Nutr. 2013, 53, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Chhunchha, B.; Fatma, N.; Kubo, E.; Rai, P.; Singh, S.P.; Singh, D.P. Curcumin abates hypoxia-induced oxidative stress based-er stress-mediated cell death in mouse hippocampal cells (ht22) by controlling prdx6 and nf-kappab regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C636–C655. [Google Scholar] [CrossRef] [PubMed]
- Niimi, K.; Yasui, T.; Hirose, M.; Hamamoto, S.; Itoh, Y.; Okada, A.; Kubota, Y.; Kojima, Y.; Tozawa, K.; Sasaki, S.; et al. Mitochondrial permeability transition pore opening induces the initial process of renal calcium crystallization. Free Radic. Biol. Med. 2012, 52, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis Int. J. Program. Cell Death 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Biasi, F.; Leonarduzzi, G. 4-hydroxynonenal-protein adducts: A reliable biomarker of lipid oxidation in liver diseases. Mol. Asp. Med. 2008, 29, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Picardo, S.L.; Maher, S.G.; O’Sullivan, J.N.; Reynolds, J.V. Barrett’s to oesophageal cancer sequence: A model of inflammatory-driven upper gastrointestinal cancer. Dig. Surg. 2012, 29, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Polkowski, W.; van Lanschot, J.J.; Ten Kate, F.J.; Baak, J.P.; Tytgat, G.N.; Obertop, H.; Voorn, W.J.; Offerhaus, G.J. The value of p53 and ki67 as markers for tumour progression in the barrett’s dysplasia-carcinoma sequence. Surg. Oncol. 1995, 4, 163–171. [Google Scholar] [CrossRef]
- Nakamura, K.; Miura, D.; Kusano, K.F.; Fujimoto, Y.; Sumita-Yoshikawa, W.; Fuke, S.; Nishii, N.; Nagase, S.; Hata, Y.; Morita, H.; et al. 4-hydroxy-2-nonenal induces calcium overload via the generation of reactive oxygen species in isolated rat cardiac myocytes. J. Card. Fail. 2009, 15, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.T.; Biniecka, M.; Kennedy, A.; McCormick, J.; Fitzgerald, O.; Bresnihan, B.; Buggy, D.; Taylor, C.T.; O’Sullivan, J.; Fearon, U.; et al. Synovial tissue hypoxia and inflammation in vivo. Ann. Rheum. Dis. 2010, 69, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, F.; Schneider, P.M.; Metzger, R.; Warnecke-Eberz, U.; Baldus, S.E.; Dienes, H.P.; Aikou, T.; Hoelscher, A.H. Mutations in the mitochondrial DNA d-loop region occur frequently in adenocarcinoma in barrett’s esophagus. Oncogene 2002, 21, 3780–3783. [Google Scholar] [CrossRef] [PubMed]
- Picardo, S.L.; O’Brien, M.P.; Feighery, R.; O’Toole, D.; Ravi, N.; O’Farrell, N.J.; O’Sullivan, J.N.; Reynolds, J.V. A barrett’s esophagus registry of over 1000 patients from a specialist center highlights greater risk of progression than population-based registries and high risk of low grade dysplasia. Dis. Esophagus 2014, 28, 121–126. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Farrell, N.J.; Phelan, J.J.; Feighery, R.; Doyle, B.; Picardo, S.L.; Ravi, N.; O’Toole, D.; Reynolds, J.V.; O’Sullivan, J. Differential Expression Profiles of Oxidative Stress Levels, 8-oxo-dG and 4-HNE, in Barrett’s Esophagus Compared to Esophageal Adenocarcinoma. Int. J. Mol. Sci. 2019, 20, 4449. https://doi.org/10.3390/ijms20184449
O’Farrell NJ, Phelan JJ, Feighery R, Doyle B, Picardo SL, Ravi N, O’Toole D, Reynolds JV, O’Sullivan J. Differential Expression Profiles of Oxidative Stress Levels, 8-oxo-dG and 4-HNE, in Barrett’s Esophagus Compared to Esophageal Adenocarcinoma. International Journal of Molecular Sciences. 2019; 20(18):4449. https://doi.org/10.3390/ijms20184449
Chicago/Turabian StyleO’Farrell, Naoimh J., James J. Phelan, Ronan Feighery, Brendan Doyle, Sarah L. Picardo, Narayanasamy Ravi, Dermot O’Toole, John V. Reynolds, and Jacintha O’Sullivan. 2019. "Differential Expression Profiles of Oxidative Stress Levels, 8-oxo-dG and 4-HNE, in Barrett’s Esophagus Compared to Esophageal Adenocarcinoma" International Journal of Molecular Sciences 20, no. 18: 4449. https://doi.org/10.3390/ijms20184449
APA StyleO’Farrell, N. J., Phelan, J. J., Feighery, R., Doyle, B., Picardo, S. L., Ravi, N., O’Toole, D., Reynolds, J. V., & O’Sullivan, J. (2019). Differential Expression Profiles of Oxidative Stress Levels, 8-oxo-dG and 4-HNE, in Barrett’s Esophagus Compared to Esophageal Adenocarcinoma. International Journal of Molecular Sciences, 20(18), 4449. https://doi.org/10.3390/ijms20184449