Chemoresistance in Pancreatic Cancer

 , , and
, , and
Abstract
:1. Introduction
2. Current Treatment Options of Pancreatic Cancer and Limitations
3. Chemoresistance in Pancreatic Cancer
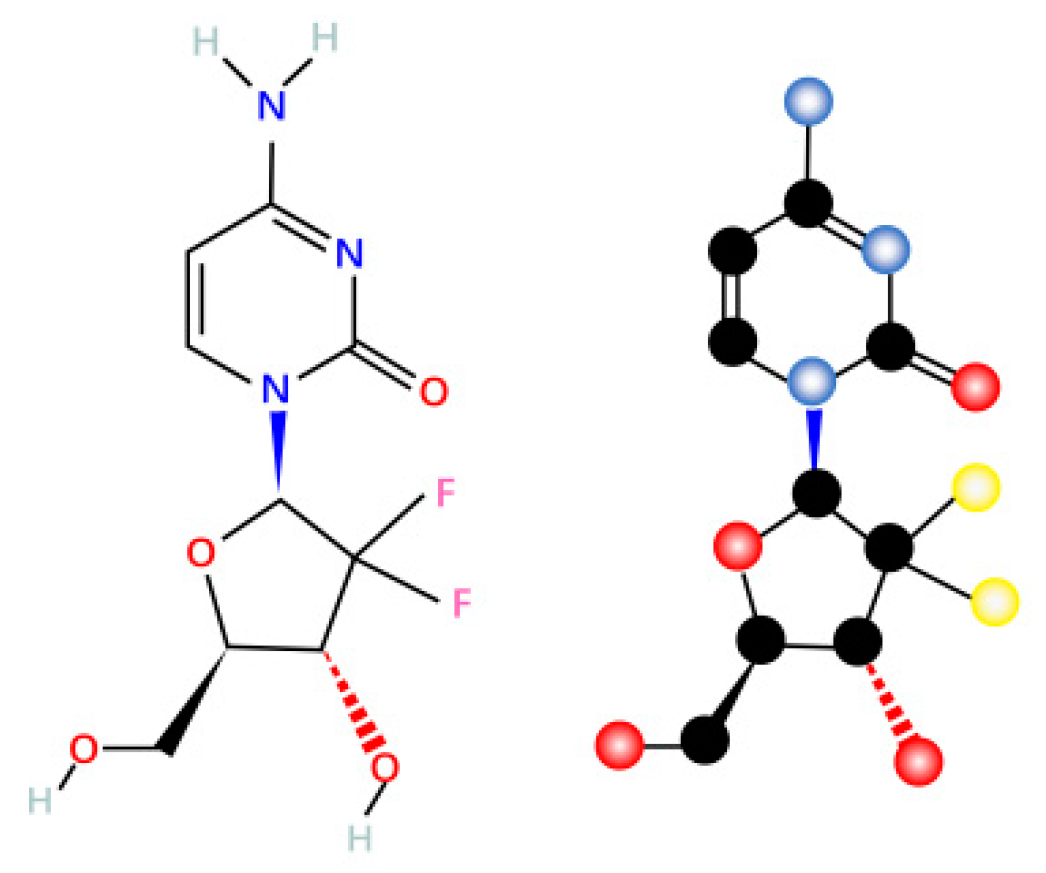
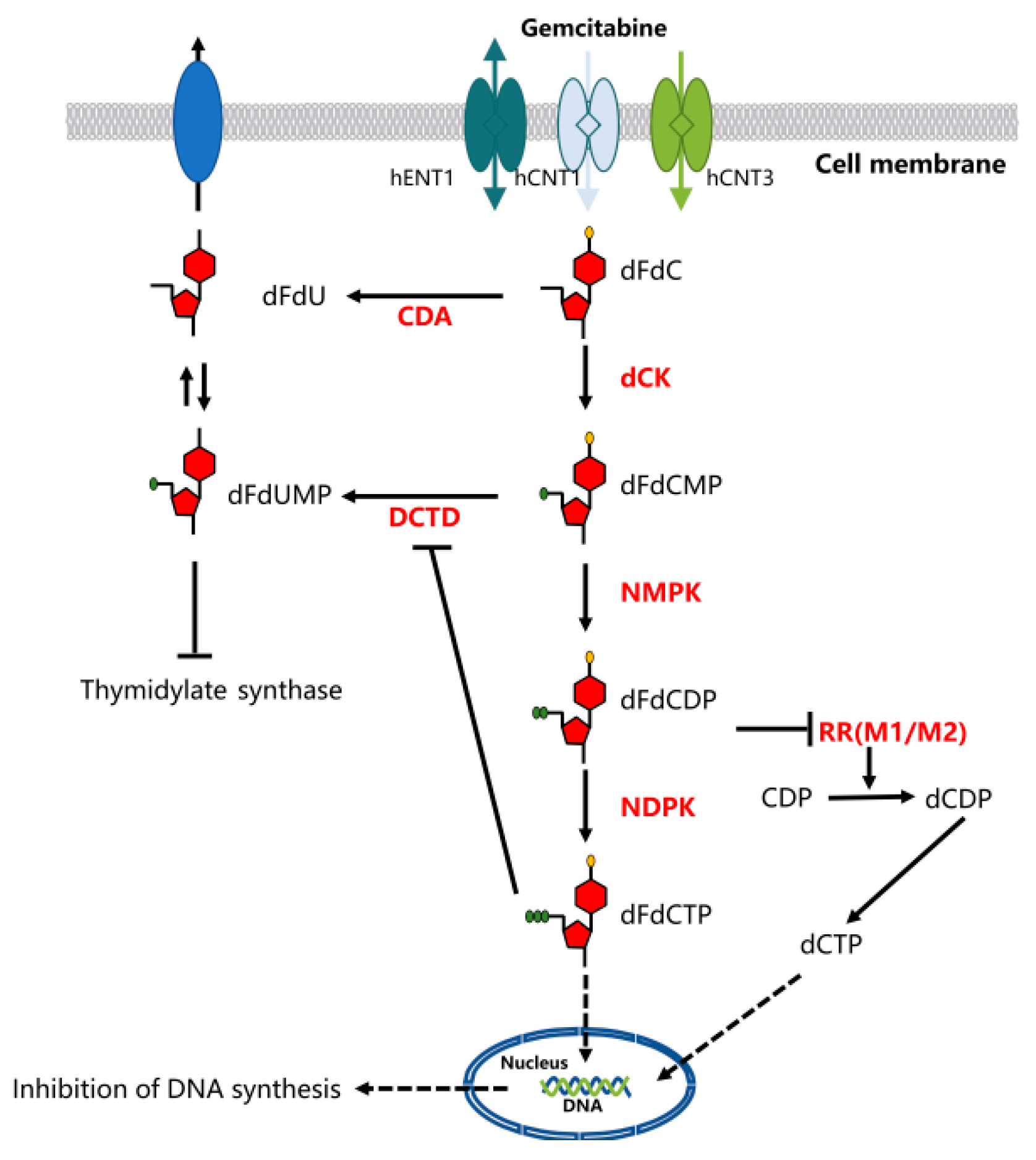
4. Mechanism of Action of Gemcitabine
5. Gemcitabine Resistance Mechanisms
5.1. Nucleoside Transporters
5.2. Nucleoside Enzymes
5.3. Epithelial–Mesenchymal Transition
5.4. Microenvironmental Factors
5.5. Impact of Other Relevant Factors
6. Potential Ways to Improve Gemcitabine Uptake and Efficacy
7. Genome-Scale Screening Drug Resistant Genes
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| CAFs | cancer-related fibroblasts |
| dCK | deoxycytidine kinase |
| dCTP | deoxycytidine triphosphate |
| dFdC | 2′,2′-difluoro-2′-deoxycytidine |
| ECM | extracellular matrix |
| EMT | epithelial-mesenchymal transition |
| MSCs | mesenchymal stem cells |
| NT | nucleoside transporter |
| OS | overall survival |
| PDAC | pancreatic ductal adenocarcinoma |
| PSCs | pancreatic stellate cells |
| RR | ribonucleotide reductase |
| TS | thymidylate synthase |
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Smith, L.M.; Patel, A.; Menning, M.; Watley, D.C.; Malik, S.S.; Krishn, S.R.; Mallya, K.; Aithal, A.; Sasson, A.R.; et al. A Combination of MUC5AC and CA19-9 Improves the Diagnosis of Pancreatic Cancer: A Multicenter Study. Am. J. Gastroenterol. 2017, 112, 172–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Baine, M.J.; Jain, M.; Sasson, A.R.; Batra, S.K. Early diagnosis of pancreatic cancer: Challenges and new developments. Biomark. Med. 2012, 6, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Dumont, R.; Puleo, F.; Collignon, J.; Meurisse, N.; Chavez, M.; Seidel, L.; Gast, P.; Polus, M.; Loly, C.; Delvenne, P.; et al. A single center experience in resectable pancreatic ductal adenocarcinoma: The limitations of the surgery-first approach. Critical review of the literature and proposals for practice update. Acta Gastroenterol. Belg. 2017, 80, 451–461. [Google Scholar]
- Labori, K.J.; Katz, M.H.; Tzeng, C.W.; Bjornbeth, B.A.; Cvancarova, M.; Edwin, B.; Kure, E.H.; Eide, T.J.; Dueland, S.; Buanes, T.; et al. Impact of early disease progression and surgical complications on adjuvant chemotherapy completion rates and survival in patients undergoing the surgery first approach for resectable pancreatic ductal adenocarcinoma—A population-based cohort study. Acta Oncol. 2016, 55, 265–277. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Hwang, I.; Yoo, C.; Kim, K.P.; Jeong, J.H.; Chang, H.M.; Lee, S.S.; Park, D.H.; Song, T.J.; Seo, D.W.; et al. Nab-paclitaxel plus gemcitabine versus FOLFIRINOX as the first-line chemotherapy for patients with metastatic pancreatic cancer: Retrospective analysis. Investig. New Drugs 2018, 36, 732–741. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.; Bonafede, M.; Cai, Q.; Princic, N.; Tran, O.; Pelletier, C.; Parisi, M.; Patel, M. Comparison of treatment patterns and economic outcomes among metastatic pancreatic cancer patients initiated on nab-paclitaxel plus gemcitabine versus FOLFIRINOX. Expert Rev. Clin. Pharmacol. 2017, 10, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, R.D.; Ho, M.; Renouf, D.J.; Lim, H.J.; Gill, S.; Ruan, J.Y.; Cheung, W.Y. Eligibility of Metastatic Pancreatic Cancer Patients for First-Line Palliative Intent nab-Paclitaxel Plus Gemcitabine Versus FOLFIRINOX. Am. J. Clin. Oncol. 2017, 40, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Nomura, A.; Saluja, A.; Banerjee, S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology 2017, 17, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17, v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2′,2′-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22, 3–10. [Google Scholar] [PubMed]
- Rauchwerger, D.R.; Firby, P.S.; Hedley, D.W.; Moore, M.J. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Res. 2000, 60, 6075–6079. [Google Scholar] [PubMed]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: A mechanism of self-potentiation. Cancer Res. 1992, 52, 533–539. [Google Scholar] [PubMed]
- Kelderman, S.; Schumacher, T.N.; Haanen, J.B. Acquired and intrinsic resistance in cancer immunotherapy. Mol. Oncol. 2014, 8, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.M.; Pinedo, H.M.; Peters, G.J. Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine). Drug Resist. Updat. 2002, 5, 19–33. [Google Scholar] [CrossRef]
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar] [PubMed]
- Nordh, S.; Ansari, D.; Andersson, R. hENT1 expression is predictive of gemcitabine outcome in pancreatic cancer: A systematic review. World J. Gastroenterol. 2014, 20, 8482–8490. [Google Scholar] [CrossRef] [PubMed]
- Elebro, J.; Ben Dror, L.; Heby, M.; Nodin, B.; Jirstrom, K.; Eberhard, J. Prognostic effect of hENT1, dCK and HuR expression by morphological type in periampullary adenocarcinoma, including pancreatic cancer. Acta Oncol. 2016, 55, 286–296. [Google Scholar] [CrossRef]
- Orlandi, A.; Calegari, M.A.; Martini, M.; Cocomazzi, A.; Bagala, C.; Indellicati, G.; Zurlo, V.; Basso, M.; Cassano, A.; Larocca, L.M.; et al. Gemcitabine versus FOLFIRINOX in patients with advanced pancreatic adenocarcinoma hENT1-positive: Everything was not too bad back when everything seemed worse. Clin. Transl. Oncol. 2016, 18, 988–995. [Google Scholar] [CrossRef]
- Bird, N.T.; Elmasry, M.; Jones, R.; Psarelli, E.; Dodd, J.; Malik, H.; Greenhalf, W.; Kitteringham, N.; Ghaneh, P.; Neoptolemos, J.P.; et al. Immunohistochemical hENT1 expression as a prognostic biomarker in patients with resected pancreatic ductal adenocarcinoma undergoing adjuvant gemcitabine-based chemotherapy. Br. J. Surg. 2017, 104, 328–336. [Google Scholar] [CrossRef]
- Elander, N.O.; Aughton, K.; Ghaneh, P.; Neoptolemos, J.P.; Palmer, D.H.; Cox, T.F.; Campbell, F.; Costello, E.; Halloran, C.M.; Mackey, J.R.; et al. Expression of dihydropyrimidine dehydrogenase (DPD) and hENT1 predicts survival in pancreatic cancer. Br. J. Cancer 2018, 118, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Nakanishi, T.; Tajima, H.; Yamazaki, M.; Yokono, R.; Takabayashi, M.; Shimada, T.; Sawamoto, K.; Miyamoto, K.; Kitagawa, H.; et al. Saturable Hepatic Extraction of Gemcitabine Involves Biphasic Uptake Mediated by Nucleoside Transporters Equilibrative Nucleoside Transporter 1 and 2. J. Pharm. Sci. 2015, 104, 3162–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boswell-Casteel, R.C.; Hays, F.A. Equilibrative nucleoside transporters—A review. Nucleotides Nucleic Acids 2017, 36, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Mohelnikova-Duchonova, B.; Brynychova, V.; Hlavac, V.; Kocik, M.; Oliverius, M.; Hlavsa, J.; Honsova, E.; Mazanec, J.; Kala, Z.; Melichar, B.; et al. The association between the expression of solute carrier transporters and the prognosis of pancreatic cancer. Cancer Chemother. Pharmacol. 2013, 72, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torras, S.; Vidal-Pla, A.; Cano-Soldado, P.; Huber-Ruano, I.; Mazo, A.; Pastor-Anglada, M. Concentrative nucleoside transporter 1 (hCNT1) promotes phenotypic changes relevant to tumor biology in a translocation-independent manner. Cell Death Dis. 2013, 4, e648. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Hung, S.W.; Patel, B.; Lovin, D.; Govindarajan, R. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Res. 2011, 71, 1825–1835. [Google Scholar] [CrossRef]
- Saiki, Y.; Yoshino, Y.; Fujimura, H.; Manabe, T.; Kudo, Y.; Shimada, M.; Mano, N.; Nakano, T.; Lee, Y.; Shimizu, S.; et al. DCK is frequently inactivated in acquired gemcitabine-resistant human cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 98–104. [Google Scholar] [CrossRef]
- Ohhashi, S.; Ohuchida, K.; Mizumoto, K.; Fujita, H.; Egami, T.; Yu, J.; Toma, H.; Sadatomi, S.; Nagai, E.; Tanaka, M. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer Res. 2008, 28, 2205–2212. [Google Scholar]
- Funamizu, N.; Okamoto, A.; Kamata, Y.; Misawa, T.; Uwagawa, T.; Gocho, T.; Yanaga, K.; Manome, Y. Is the resistance of gemcitabine for pancreatic cancer settled only by overexpression of deoxycytidine kinase? Oncol. Rep. 2010, 23, 471–475. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Z.; Bai, Z.; Ma, X.; Guo, W.; Wang, Y. Enhancement of gemcitabine sensitivity in pancreatic cancer by co-regulation of dCK and p8 expression. Oncol. Rep. 2011, 25, 963–970. [Google Scholar] [CrossRef]
- Ohmine, K.; Kawaguchi, K.; Ohtsuki, S.; Motoi, F.; Ohtsuka, H.; Kamiie, J.; Abe, T.; Unno, M.; Terasaki, T. Quantitative Targeted Proteomics of Pancreatic Cancer: Deoxycytidine Kinase Protein Level Correlates to Progression-Free Survival of Patients Receiving Gemcitabine Treatment. Mol. Pharm. 2015, 12, 3282–3291. [Google Scholar] [CrossRef]
- Sierzega, M.; Pach, R.; Kulig, P.; Legutko, J.; Kulig, J. Prognostic Implications of Expression Profiling for Gemcitabine-Related Genes (hENT1, dCK, RRM1, RRM2) in Patients with Resectable Pancreatic Adenocarcinoma Receiving Adjuvant Chemotherapy. Pancreas 2017, 46, 684–689. [Google Scholar] [CrossRef] [PubMed]
- McAllister, F.; Pineda, D.M.; Jimbo, M.; Lal, S.; Burkhart, R.A.; Moughan, J.; Winter, K.A.; Abdelmohsen, K.; Gorospe, M.; Acosta Ade, J.; et al. dCK expression correlates with 5-fluorouracil efficacy and HuR cytoplasmic expression in pancreatic cancer: A dual-institutional follow-up with the RTOG 9704 trial. Cancer Biol. Ther. 2014, 15, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Costantino, C.L.; Witkiewicz, A.K.; Kuwano, Y.; Cozzitorto, J.A.; Kennedy, E.P.; Dasgupta, A.; Keen, J.C.; Yeo, C.J.; Gorospe, M.; Brody, J.R. The role of HuR in gemcitabine efficacy in pancreatic cancer: HuR Up-regulates the expression of the gemcitabine metabolizing enzyme deoxycytidine kinase. Cancer Res. 2009, 69, 4567–4572. [Google Scholar] [CrossRef] [PubMed]
- Tatarian, T.; Jiang, W.; Leiby, B.E.; Grigoli, A.; Jimbo, M.; Dabbish, N.; Neoptolemos, J.P.; Greenhalf, W.; Costello, E.; Ghaneh, P.; et al. Cytoplasmic HuR Status Predicts Disease-free Survival in Resected Pancreatic Cancer: A Post-hoc Analysis from the International Phase III ESPAC-3 Clinical Trial. Ann. Surg. 2018, 267, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.L.; Zhou, Y.H.; Lyu, Y.; Yan, H.; Dai, G.H. Effect of ribonucleotide reductase M1 expression on overall survival in patients with pancreatic cancer receiving gemcitabine chemotherapy: A literature-based meta-analysis. J. Clin. Pharm. Ther. 2018, 43, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, T.; Miyagi, Y.; Murakawa, M.; Yamaoku, K.; Atsumi, Y.; Shiozawa, M.; Ueno, M.; Morimoto, M.; Oshima, T.; Yukawa, N.; et al. Clinical implications of ribonucleotide reductase subunit M1 in patients with pancreatic cancer who undergo curative resection followed by adjuvant chemotherapy with gemcitabine. Oncol. Lett. 2017, 13, 3423–3430. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, W.; Fu, M.; Yang, A.; Huang, H.; Xie, J. Establishment of human pancreatic cancer gemcitabineresistant cell line with ribonucleotide reductase overexpression. Oncol. Rep. 2015, 33, 383–390. [Google Scholar] [CrossRef]
- Nakahira, S.; Nakamori, S.; Tsujie, M.; Takahashi, Y.; Okami, J.; Yoshioka, S.; Yamasaki, M.; Marubashi, S.; Takemasa, I.; Miyamoto, A.; et al. Involvement of ribonucleotide reductase M1 subunit overexpression in gemcitabine resistance of human pancreatic cancer. Int. J. Cancer 2007, 120, 1355–1363. [Google Scholar] [CrossRef]
- Goan, Y.G.; Zhou, B.; Hu, E.; Mi, S.; Yen, Y. Overexpression of ribonucleotide reductase as a mechanism of resistance to 2,2-difluorodeoxycytidine in the human KB cancer cell line. Cancer Res. 1999, 59, 4204–4207. [Google Scholar]
- Minami, K.; Shinsato, Y.; Yamamoto, M.; Takahashi, H.; Zhang, S.; Nishizawa, Y.; Tabata, S.; Ikeda, R.; Kawahara, K.; Tsujikawa, K.; et al. Ribonucleotide reductase is an effective target to overcome gemcitabine resistance in gemcitabine-resistant pancreatic cancer cells with dual resistant factors. J. Pharmacol. Sci. 2015, 127, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Liu, H.; Lu, Y.; Zhao, X.; Fan, D. Epithelial-to-Mesenchymal Transition: Liaison between Cancer Metastasis and Drug Resistance. Crit. Rev. Oncog. 2017, 22, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Lambies, G.; Miceli, M.; Martinez-Guillamon, C. TGFbeta-Activated USP27X Deubiquitinase Regulates Cell Migration and Chemoresistance via Stabilization of Snail1. Cancer Res. 2019, 79, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Galvan, J.A.; Zlobec, I.; Wartenberg, M.; Lugli, A.; Gloor, B.; Perren, A.; Karamitopoulou, E. Expression of E-cadherin repressors SNAIL, ZEB1 and ZEB2 by tumour and stromal cells influences tumour-budding phenotype and suggests heterogeneity of stromal cells in pancreatic cancer. Br. J. Cancer 2015, 112, 1944–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, R.T.; Veronese, N. Prognostic Role of High-Grade Tumor Budding in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis with a Focus on Epithelial to Mesenchymal Transition. Cancers 2019, 11, 113. [Google Scholar] [CrossRef]
- Chouat, E.; Zehani, A.; Chelly, I.; Njima, M.; Maghrebi, H.; Bani, M.A.; Njim, L.; Zakhama, A.; Haouet, S.; Kchir, N. Tumor budding is a prognostic factor linked to epithelial mesenchymal transition in pancreatic ductal adenocarcinoma. Study report and literature review. Pancreatology 2018, 18, 79–84. [Google Scholar] [CrossRef]
- Kohler, I.; Bronsert, P.; Timme, S.; Werner, M.; Brabletz, T.; Hopt, U.T.; Schilling, O.; Bausch, D.; Keck, T.; Wellner, U.F. Detailed analysis of epithelial-mesenchymal transition and tumor budding identifies predictors of long-term survival in pancreatic ductal adenocarcinoma. J. Gastroenterol. Hepatol. 2015, 30, 78–84. [Google Scholar] [CrossRef]
- Apte, M.V.; Xu, Z.; Pothula, S.; Goldstein, D.; Pirola, R.C.; Wilson, J.S. Pancreatic cancer: The microenvironment needs attention too! Pancreatology 2015, 15, S32–S38. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D.; Black, B.S.; Sorenson, H.L.; Knowlton, A.E.; Thomas, R.M.; Sarosi, G.A.; Moldawer, L.L.; Behrns, K.E.; Liu, C.; George, T.J.; et al. The inflammatory milieu within the pancreatic cancer microenvironment correlates with clinicopathologic parameters, chemoresistance and survival. BMC Cancer 2015, 15, 783. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Li, J.; Sun, H.; Liu, S.; Cui, Y.; Li, F. HES 1 is essential for chemoresistance induced by stellate cells and is associated with poor prognosis in pancreatic cancer. Oncol. Rep. 2015, 33, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, H.; Guan, J.; Wang, L.; Ren, X.; Shi, X.; Liang, Z.; Liu, T. Paracrine SDF-1alpha signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget 2015, 6, 3085–3097. [Google Scholar] [CrossRef]
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour. Biol. 2016, 37, 15283–15291. [Google Scholar] [CrossRef]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef]
- Tostar, U.; Toftgard, R.; Zaphiropoulos, P.G.; Shimokawa, T. Reduction of human embryonal rhabdomyosarcoma tumor growth by inhibition of the hedgehog signaling pathway. Genes Cancer 2010, 1, 941–951. [Google Scholar] [CrossRef]
- Hui, M.; Cazet, A.; Nair, R.; Watkins, D.N.; O’Toole, S.A.; Swarbrick, A. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy. Breast Cancer Res. 2013, 15, 203. [Google Scholar] [CrossRef]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Giroux-Leprieur, E.; Costantini, A.; Ding, V.W.; He, B. Hedgehog Signaling in Lung Cancer: From Oncogenesis to Cancer Treatment Resistance. Int. J. Mol. Sci. 2018, 19, 2835. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.T.; Zhuan-Sun, Y.X.; Zhuang, Y.Y.; Wei, S.L.; Tang, J.; Chen, W.B.; Zhang, S.N. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int. J. Oncol. 2012, 41, 1707–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef]
- Duluc, C.; Moatassim-Billah, S.; Chalabi-Dchar, M.; Perraud, A.; Samain, R.; Breibach, F.; Gayral, M.; Cordelier, P.; Delisle, M.B.; Bousquet-Dubouch, M.P.; et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol. Med. 2015, 7, 735–753. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-Stroma IL1beta-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712. [Google Scholar] [CrossRef]
- Xiong, G.; Feng, M.; Yang, G.; Zheng, S.; Song, X.; Cao, Z.; You, L.; Zheng, L.; Hu, Y.; Zhang, T.; et al. The underlying mechanisms of non-coding RNAs in the chemoresistance of pancreatic cancer. Cancer Lett. 2017, 397, 94–102. [Google Scholar] [CrossRef]
- Madurantakam Royam, M.; Ramesh, R.; Shanker, R.; Sabarimurugan, S. miRNA Predictors of Pancreatic Cancer Chemotherapeutic Response: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 900. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Wang, J.; Chen, X.; Liu, L. Role of microRNA in anticancer drug resistance. Int. J. Cancer 2010, 126, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, J.; Chen, Q.; Qu, C.; Zhang, J.; Chen, E.; Zhang, Y.; Wang, Y.; Ni, L.; Liang, T. Gemcitabine enhances OSI-027 cytotoxicity by upregulation of miR-663a in pancreatic ductal adenocarcinoma cells. Am. J. Transl. Res. 2019, 11, 473–485. [Google Scholar] [PubMed]
- Huang, L.; Hu, C.; Cao, H.; Wu, X.; Wang, R.; Lu, H.; Li, H.; Chen, H. MicroRNA-29c Increases the Chemosensitivity of Pancreatic Cancer Cells by Inhibiting USP22 Mediated Autophagy. Cell Physiol. Biochem. 2018, 47, 747–758. [Google Scholar] [CrossRef]
- Xiong, G.; Huang, H.; Feng, M.; Yang, G.; Zheng, S.; You, L.; Zheng, L.; Hu, Y.; Zhang, T.; Zhao, Y. MiR-10a-5p targets TFAP2C to promote gemcitabine resistance in pancreatic ductal adenocarcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 76. [Google Scholar] [CrossRef]
- Amponsah, P.S.; Fan, P.; Bauer, N.; Zhao, Z.; Gladkich, J.; Fellenberg, J.; Herr, I. microRNA-210 overexpression inhibits tumor growth and potentially reverses gemcitabine resistance in pancreatic cancer. Cancer Lett. 2017, 388, 107–117. [Google Scholar] [CrossRef]
- Fan, P.; Liu, L.; Yin, Y.; Zhao, Z.; Zhang, Y.; Amponsah, P.S.; Xiao, X.; Bauer, N.; Abukiwan, A.; Nwaeburu, C.C.; et al. MicroRNA-101-3p reverses gemcitabine resistance by inhibition of ribonucleotide reductase M1 in pancreatic cancer. Cancer Lett. 2016, 373, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhao, Z.; Zhou, Z.; Liu, R. Linc-ROR confers gemcitabine resistance to pancreatic cancer cells via inducing autophagy and modulating the miR-124/PTBP1/PKM2 axis. Cancer Chemother. Pharmacol. 2016, 78, 1199–1207. [Google Scholar] [CrossRef]
- Gu, J.; Wang, D.; Zhang, J.; Zhu, Y.; Li, Y.; Chen, H.; Shi, M.; Wang, X.; Shen, B.; Deng, X.; et al. GFRalpha2 prompts cell growth and chemoresistance through down-regulating tumor suppressor gene PTEN via Mir-17-5p in pancreatic cancer. Cancer Lett. 2016, 380, 434–441. [Google Scholar] [CrossRef]
- Tian, X.; Shivapurkar, N.; Wu, Z.; Hwang, J.J.; Pishvaian, M.J.; Weiner, L.M.; Ley, L.; Zhou, D.; Zhi, X.; Wellstein, A.; et al. Circulating microRNA profile predicts disease progression in patients receiving second-line treatment of lapatinib and capecitabine for metastatic pancreatic cancer. Oncol. Lett. 2016, 11, 1645–1650. [Google Scholar] [CrossRef]
- Ren, Z.G.; Dong, S.X.; Han, P.; Qi, J. miR-203 promotes proliferation, migration and invasion by degrading SIK1 in pancreatic cancer. Oncol. Rep. 2016, 35, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Gao, X.; Hu, F.; Zhang, Y.; Gou, X. SLFN11 is a general target for enhancing the sensitivity of cancer to chemotherapy (DNA-damaging agents). J. Drug Target 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Thomas, A.; Miettinen, M.; Pommier, Y. Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol. Ther. 2019, 201, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Tang, S.W.; Leo, E.; Baechler, S.A.; Redon, C.E.; Zhang, H.; Al Abo, M.; Rajapakse, V.N.; Nakamura, E.; Jenkins, L.M.M.; et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 2018, 69, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Feng, Y.; Yu, G.K.; Ru, Y.; Tang, S.W.; Shen, Y.; Pommier, Y. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget 2016, 7, 76534–76550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lok, B.H.; Gardner, E.E.; Schneeberger, V.E.; Ni, A.; Desmeules, P.; Rekhtman, N.; de Stanchina, E.; Teicher, B.A.; Riaz, N.; Powell, S.N.; et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res. 2017, 23, 523–535. [Google Scholar] [CrossRef]
- Tang, S.W.; Thomas, A.; Murai, J.; Trepel, J.B.; Bates, S.E.; Rajapakse, V.N.; Pommier, Y. Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors. Clin. Cancer Res. 2018, 24, 1944–1953. [Google Scholar] [CrossRef]
- Nogales, V.; Reinhold, W.C.; Varma, S.; Martinez-Cardus, A.; Moutinho, C.; Moran, S.; Heyn, H.; Sebio, A.; Barnadas, A.; Pommier, Y.; et al. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget 2016, 7, 3084–3097. [Google Scholar] [CrossRef]
- Pietanza, M.C.; Waqar, S.N.; Krug, L.M.; Dowlati, A.; Hann, C.L.; Chiappori, A.; Owonikoko, T.K.; Woo, K.M.; Cardnell, R.J.; Fujimoto, J.; et al. Randomized, Double-Blind, Phase II Study of Temozolomide in Combination with Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2386–2394. [Google Scholar] [CrossRef]
- Skrypek, N.; Duchene, B.; Hebbar, M.; Leteurtre, E.; van Seuningen, I.; Jonckheere, N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene 2013, 32, 1714–1723. [Google Scholar] [CrossRef]
- Skrypek, N.; Vasseur, R.; Vincent, A.; Duchene, B.; Van Seuningen, I.; Jonckheere, N. The oncogenic receptor ErbB2 modulates gemcitabine and irinotecan/SN-38 chemoresistance of human pancreatic cancer cells via hCNT1 transporter and multidrug-resistance associated protein MRP-2. Oncotarget 2015, 6, 10853–10867. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Chien, P.Y.; Khan, A.R.; Sheikh, S.; Ali, S.M.; Ahmad, M.U.; Ahmad, I. In-vitro and in-vivo anti-cancer activity of a novel gemcitabine-cardiolipin conjugate. Anticancer Drugs 2006, 17, 53–61. [Google Scholar] [CrossRef]
- Bergman, A.M.; Adema, A.D.; Balzarini, J.; Bruheim, S.; Fichtner, I.; Noordhuis, P.; Fodstad, O.; Myhren, F.; Sandvold, M.L.; Hendriks, H.R.; et al. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Investig. New Drugs 2011, 29, 456–466. [Google Scholar] [CrossRef]
- Li, D.; Pant, S.; Ryan, D.P.; Laheru, D.; Bahary, N.; Dragovich, T.; Hosein, P.J.; Rolfe, L.; Saif, M.W.; LaValle, J.; et al. A phase II, open-label, multicenter study to evaluate the antitumor efficacy of CO-1.01 as second-line therapy for gemcitabine-refractory patients with stage IV pancreatic adenocarcinoma and negative tumor hENT1 expression. Pancreatology 2014, 14, 398–402. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, B.; Awada, A.; Evans, T.R.; Dueland, S.; Hendlisz, A.; Rasch, W.; Hernes, K.; Hagen, S.; Aamdal, S. A first-in-human phase I and pharmacokinetic study of CP-4126 (CO-101), a nucleoside analogue, in patients with advanced solid tumours. Cancer Chemother. Pharmacol. 2015, 76, 785–792. [Google Scholar] [CrossRef]
- Stuurman, F.E.; Voest, E.E.; Awada, A.; Witteveen, P.O.; Bergeland, T.; Hals, P.A.; Rasch, W.; Schellens, J.H.; Hendlisz, A. Phase I study of oral CP-4126, a gemcitabine derivative, in patients with advanced solid tumors. Investig. New Drugs 2013, 31, 959–966. [Google Scholar] [CrossRef]
- Poplin, E.; Wasan, H.; Rolfe, L.; Raponi, M.; Ikdahl, T.; Bondarenko, I.; Davidenko, I.; Bondar, V.; Garin, A.; Boeck, S.; et al. Randomized, multicenter, phase II study of CO-101 versus gemcitabine in patients with metastatic pancreatic ductal adenocarcinoma: Including a prospective evaluation of the role of hENT1 in gemcitabine or CO-101 sensitivity. J. Clin. Oncol. 2013, 31, 4453–4461. [Google Scholar] [CrossRef]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef]
- Wu, W.; Sigmond, J.; Peters, G.J.; Borch, R.F. Synthesis and biological activity of a gemcitabine phosphoramidate prodrug. J. Med. Chem. 2007, 50, 3743–3746. [Google Scholar] [CrossRef]
- Blagden, S.P.; Rizzuto, I.; Suppiah, P.; O’Shea, D.; Patel, M.; Spiers, L.; Sukumaran, A.; Bharwani, N.; Rockall, A.; Gabra, H.; et al. Anti-tumour activity of a first-in-class agent NUC-1031 in patients with advanced cancer: Results of a phase I study. Br. J. Cancer 2018, 119, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Daifuku, R.; Koratich, M.; Stackhouse, M. Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides. Pharmaceuticals 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Wang, F.; Di, Y.; Yao, L.; Yu, X.; Fu, D.; Li, J.; Jin, C. Antitumor effect of gemcitabine-loaded albumin nanoparticle on gemcitabine-resistant pancreatic cancer induced by low hENT1 expression. Int. J. Nanomed. 2018, 13, 4869–4880. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Bachet, J.B.; Hammel, P.; Desrame, J.; Meurisse, A.; Chibaudel, B.; Andre, T.; Debourdeau, P.; Dauba, J.; Lecomte, T.; Seitz, J.F.; et al. Nab-paclitaxel plus either gemcitabine or simplified leucovorin and fluorouracil as first-line therapy for metastatic pancreatic adenocarcinoma (AFUGEM GERCOR): A non-comparative, multicentre, open-label, randomised phase 2 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 337–346. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schutz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.I.; Shimokawa, T.; Hirono, S.; Kawai, M.; Sho, M.; Satoi, S.; Matsumoto, I.; Eguchi, H.; Murakami, Y.; Yamada, S.; et al. Effect of Neoadjuvant Nab-Paclitaxel plus Gemcitabine Therapy on Overall Survival in Patients with Borderline Resectable Pancreatic Cancer: A Prospective Multicenter Phase II Trial (NAC-GA Trial). Oncology 2017, 93, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Young, R.; Mainwaring, P.; Clingan, P.; Parnis, F.X.; Asghari, G.; Beale, P.; Aly, A.; Botteman, M.; Romano, A.; Ferrara, S.; et al. nab-Paclitaxel plus gemcitabine in metastatic pancreatic adenocarcinoma: Australian subset analyses of the phase III MPACT trial. Asia Pac. J. Clin. Oncol. 2018, 14, e325–e331. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Ghidini, M.; Torchio, M.; Corti, F.; Tomasello, G.; Niger, M.; Prinzi, N.; Nichetti, F. Comparative Effectiveness of Gemcitabine plus Nab-Paclitaxel and FOLFIRINOX in the First-Line Setting of Metastatic Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 484. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Ahmad, I.M.; Rachagani, S.; Banerjee, K.; Thompson, C.M.; Maurer, H.C.; Olive, K.P.; Bailey, K.L.; Britigan, B.E.; Kumar, S. Enhancing responsiveness of pancreatic cancer cells to gemcitabine treatment under hypoxia by heme oxygenase-1 inhibition. Transl. Res. 2019, 207, 56–69. [Google Scholar] [CrossRef]
- Belvedere, R.; Saggese, P.; Pessolano, E.; Memoli, D.; Bizzarro, V.; Rizzo, F. miR-196a Is Able to Restore the Aggressive Phenotype of Annexin A1 Knock-Out in Pancreatic Cancer Cells by CRISPR/Cas9 Genome Editing. Int. J. Mol. Sci. 2018, 19, 1967. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xie, H.; Liu, Y.; Xia, C.; Cun, X.; Long, Y.; Chen, X.; Deng, M.; Guo, R.; Zhang, Z.; et al. Knockdown of hypoxia-inducible factor-1 alpha by tumor targeted delivery of CRISPR/Cas9 system suppressed the metastasis of pancreatic cancer. J. Control Release 2019, 304, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Maresch, R.; Mueller, S.; Veltkamp, C.; Ollinger, R.; Friedrich, M.; Heid, I.; Steiger, K.; Weber, J.; Engleitner, T.; Barenboim, M.; et al. Multiplexed pancreatic genome engineering and cancer induction by transfection-based CRISPR/Cas9 delivery in mice. Int. J. Mol. Sci. 2016, 7, 10770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joung, J.; Konermann, S.; Gootenberg, J.S. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nature Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [Green Version]
- Sarr, A.; Bre, J.; Um, I.H.; Chan, T.H.; Mullen, P.; Harrison, D.J. Genome-scale CRISPR/Cas9 screen determines factors modulating sensitivity to ProTide NUC-1031. Sci. Rep. 2019, 9, 7643. [Google Scholar] [CrossRef]
- Kasap, C.; Elemento, O.; Kapoor, T.M. DrugTargetSeqR: A genomics- and CRISPR-Cas9-based method to analyze drug targets. Nat. Chem. Biol. 2014, 10, 626–628. [Google Scholar] [CrossRef]
- Ouyang, Q.; Liu, Y.; Tan, J.; Li, J.; Yang, D.; Zeng, F.; Huang, W.; Kong, Y.; Liu, Z.; Zhou, H.; et al. Loss of ZNF587B and SULF1 contributed to cisplatin resistance in ovarian cancer cell lines based on Genome-scale CRISPR/Cas9 screening. Am. J. Cancer Res. 2019, 9, 988–998. [Google Scholar]
- Cao, J.; Wei, J.; Yang, P.; Zhang, T.; Chen, Z.; He, F.; Wei, F.; Chen, H.; Hu, H.; Zhong, J.; et al. Genome-scale CRISPR-Cas9 knockout screening in gastrointestinal stromal tumor with Imatinib resistance. Mol. Cancer 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, J.E.; Wo, J.Y.; Ryan, D.P.; Clark, J.W.; Jiang, W.; Yeap, B.Y.; Drapek, L.C.; Ly, L.; Baglini, C.V.; Blaszkowsky, L.S.; et al. Total Neoadjuvant Therapy with FOLFIRINOX in Combination with Losartan Followed by Chemoradiotherapy for Locally Advanced Pancreatic Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Method | Target | Mechanism | References | |
|---|---|---|---|---|
| thymidylate synthase inhibitor | hENT1 | enhance hENT1 expression, enhance intracellular transport of gemcitabine | [22] | |
| inhibition of MUC4 and its membrane partner | hCNT1 and hCNT3 | enhance gemcitabine sensitivity via upregulation of hCNT1 and hCNT3 expression | [101] | |
| prodrug modification | NEO6002, gemcitabine-cardiolipin conjugate | Prodrug, bypass NTs | bypass NTs through drug modification | [103] |
| CP-4126, gemcitabine-elaidic acid conjugate | Prodrug, bypass hENT1 | transport into cancer cells independent of hENT1 levels | [104,105] | |
| NUC-1031, NUC050 gemcitabine-phosphoramidate | Prodrug, bypass NTs and dCK | bypass NTs and dCK through prodrug modification | [109,110,111,112] | |
| nanocarrier | GEM-HSA-NP | gemcitabine drug delivery based on NP | overcome various pathological and pharmacological barriers | [113] |
| GEM-NAB | gemcitabine drug delivery based on NP | overcome various pathological and pharmacological barriers | [114,115,116,117,118] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. https://doi.org/10.3390/ijms20184504
Zeng S, Pöttler M, Lan B, Grützmann R, Pilarsky C, Yang H. Chemoresistance in Pancreatic Cancer. International Journal of Molecular Sciences. 2019; 20(18):4504. https://doi.org/10.3390/ijms20184504
Chicago/Turabian StyleZeng, Siyuan, Marina Pöttler, Bin Lan, Robert Grützmann, Christian Pilarsky, and Hai Yang. 2019. "Chemoresistance in Pancreatic Cancer" International Journal of Molecular Sciences 20, no. 18: 4504. https://doi.org/10.3390/ijms20184504
APA StyleZeng, S., Pöttler, M., Lan, B., Grützmann, R., Pilarsky, C., & Yang, H. (2019). Chemoresistance in Pancreatic Cancer. International Journal of Molecular Sciences, 20(18), 4504. https://doi.org/10.3390/ijms20184504