Cellular Uptake and Clearance of Oxidatively-modified Apolipoprotein E3 by Cerebral Cortex Endothelial Cells

Abstract
1. Introduction
2. Results
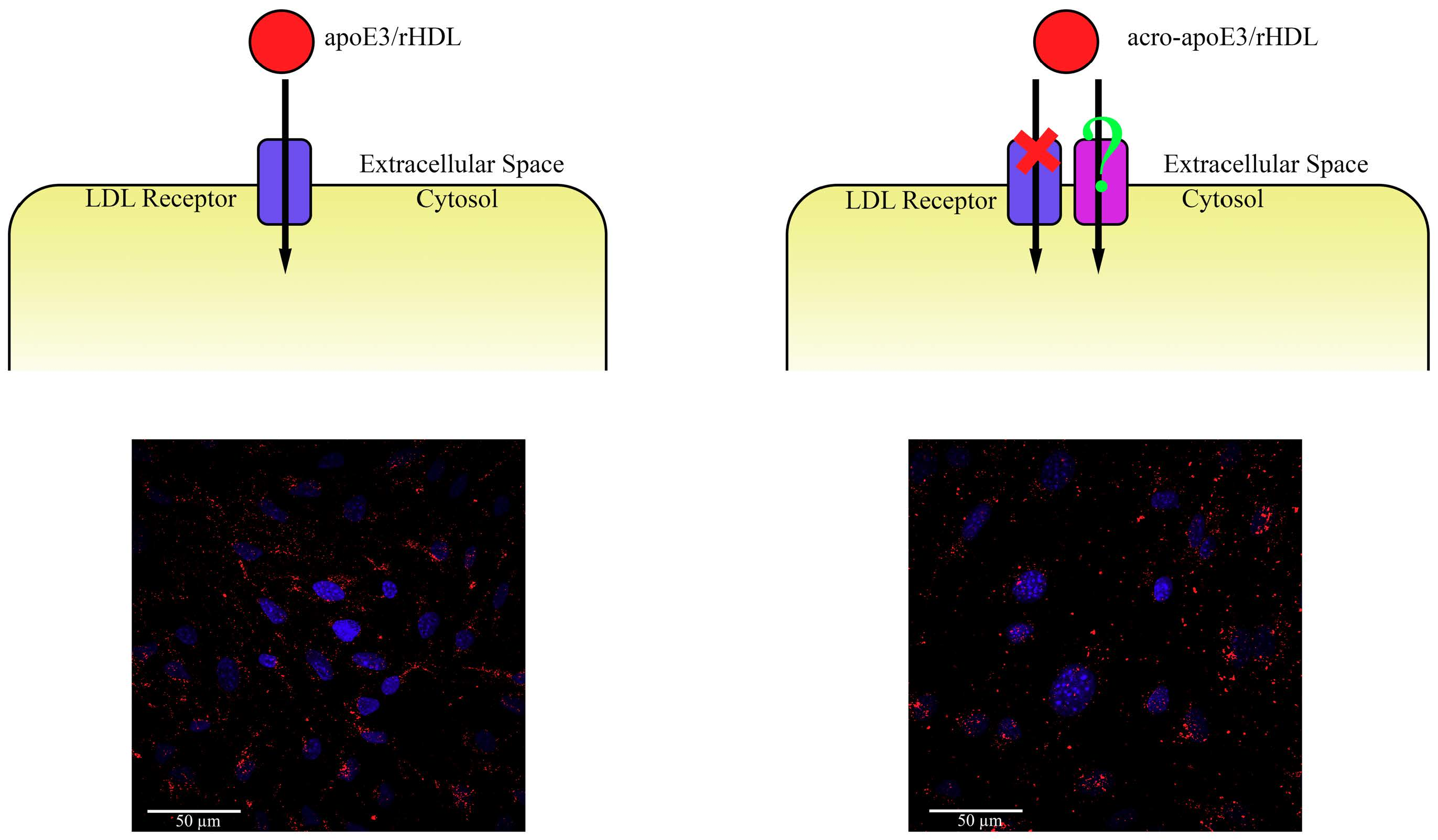
2.1. Acrolein Modification of apoE3
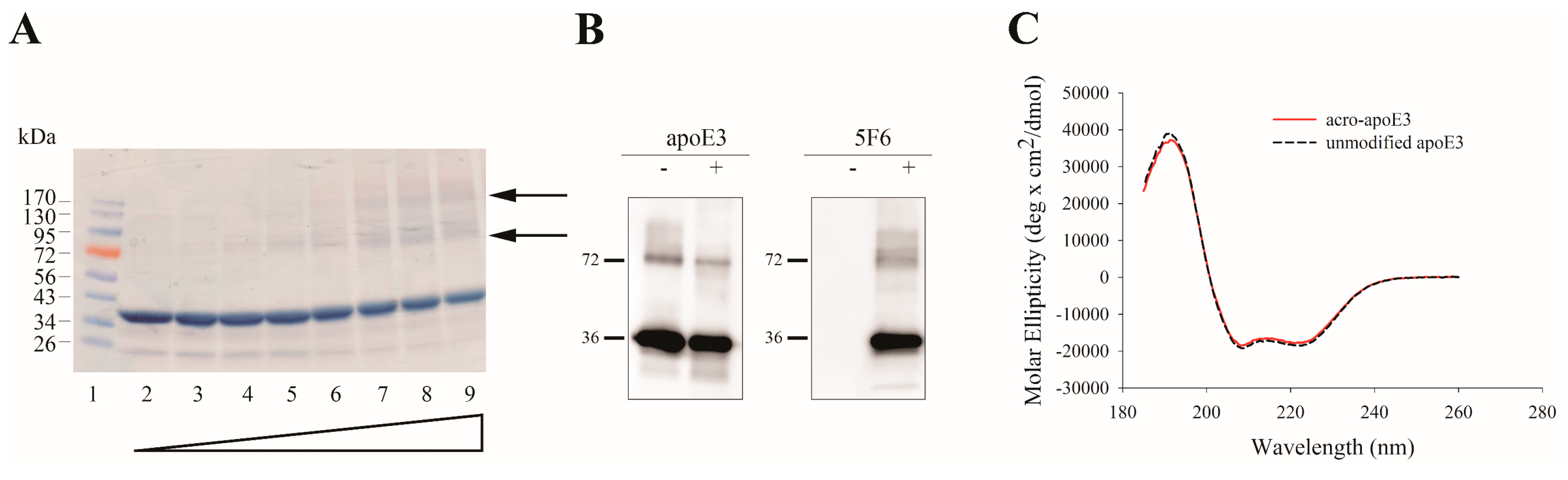
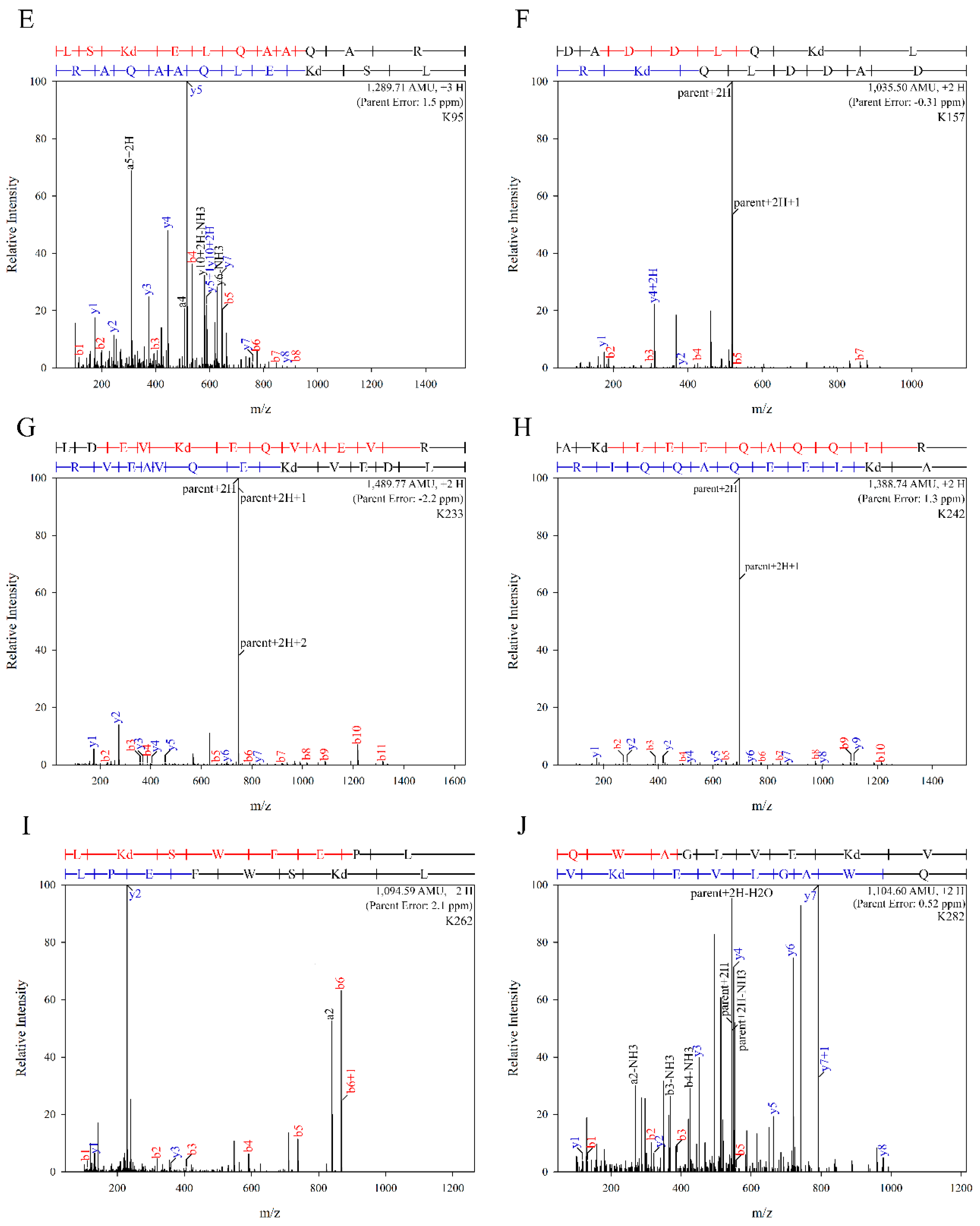
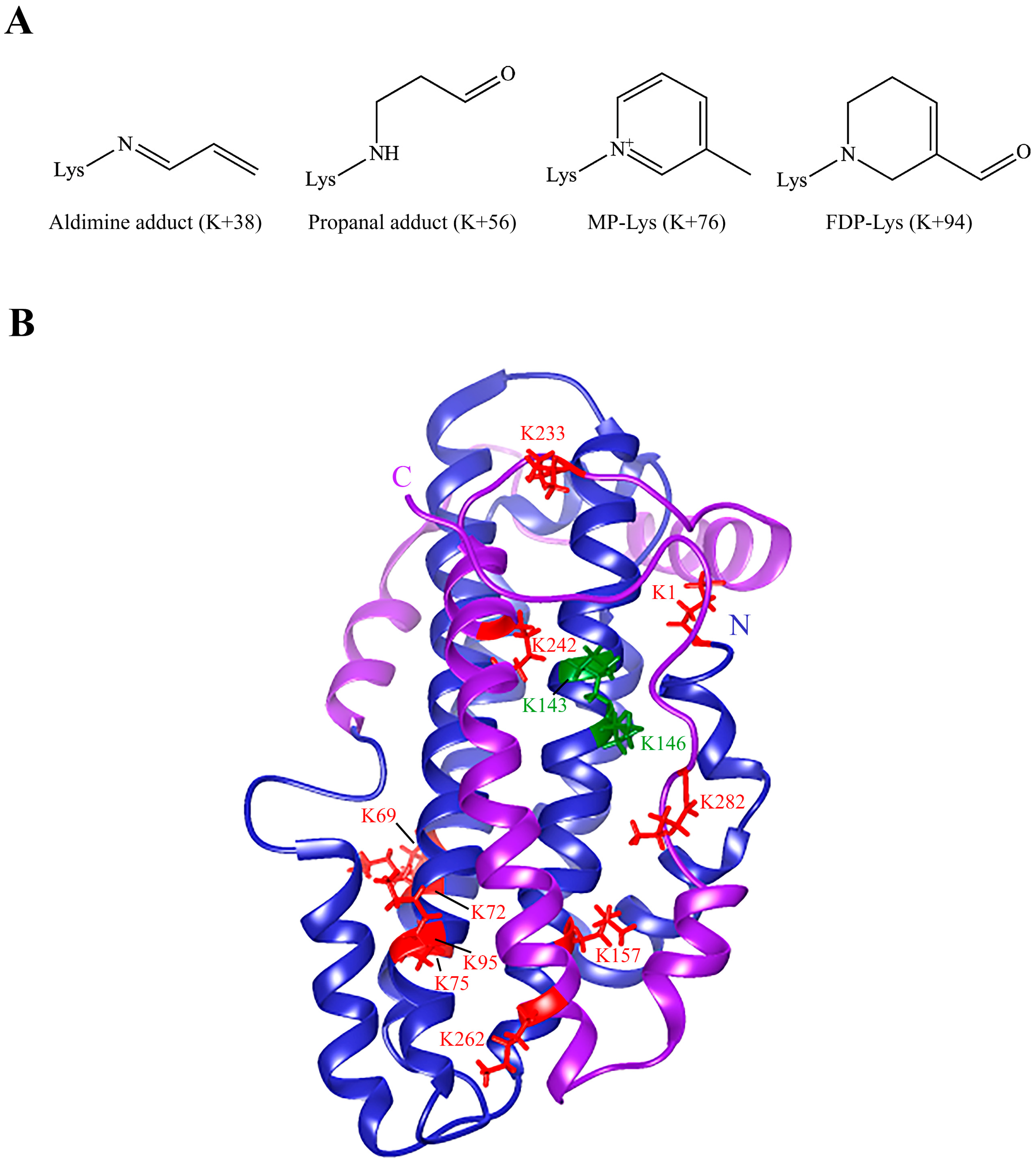
2.2. Mass Spectral Analysis
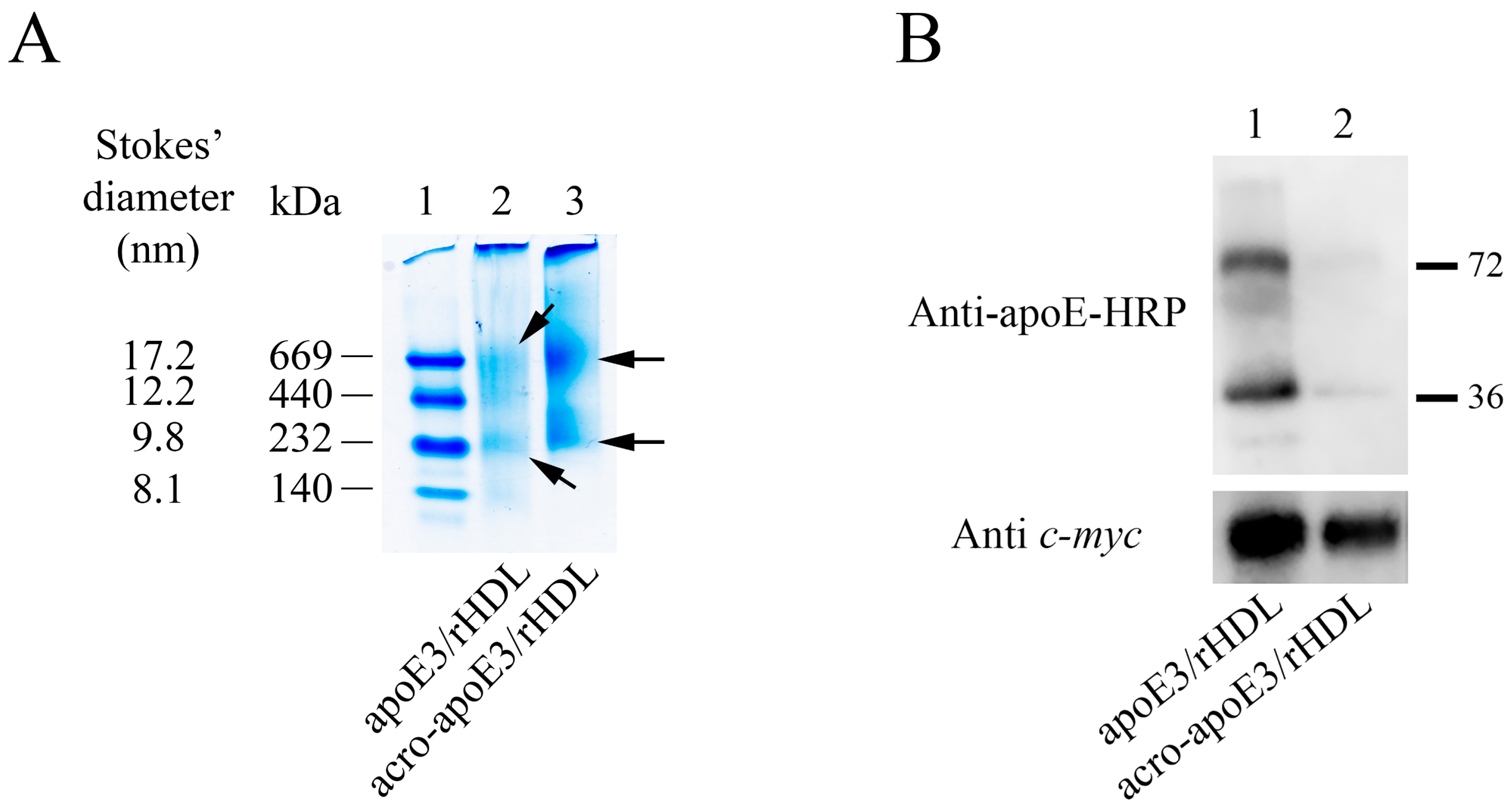
2.3. LDLr Binding Ability of acro-apoE3/rHDL
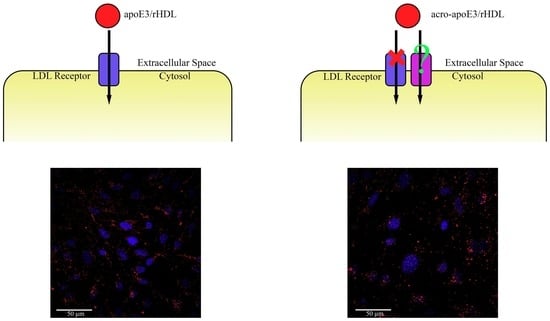
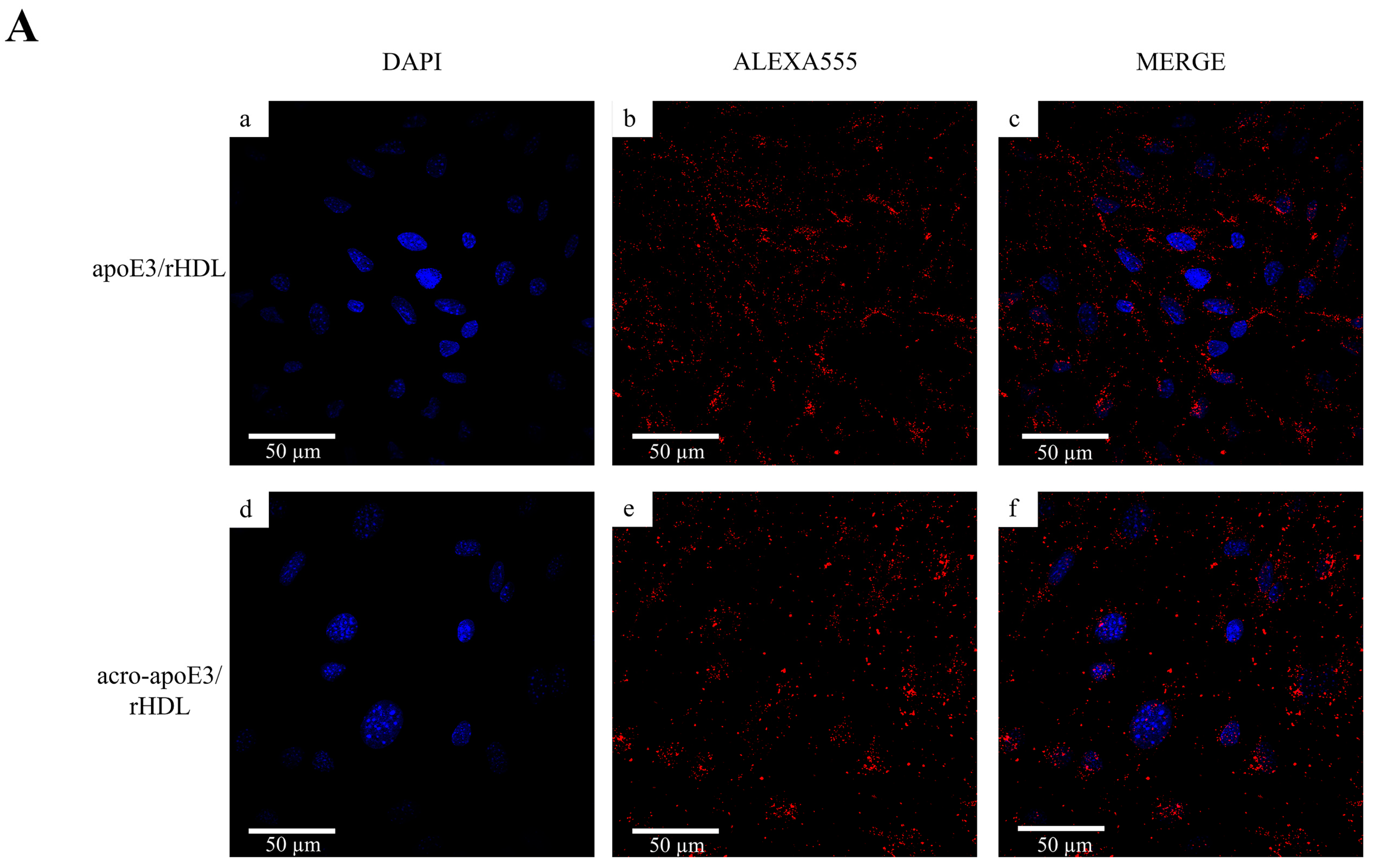
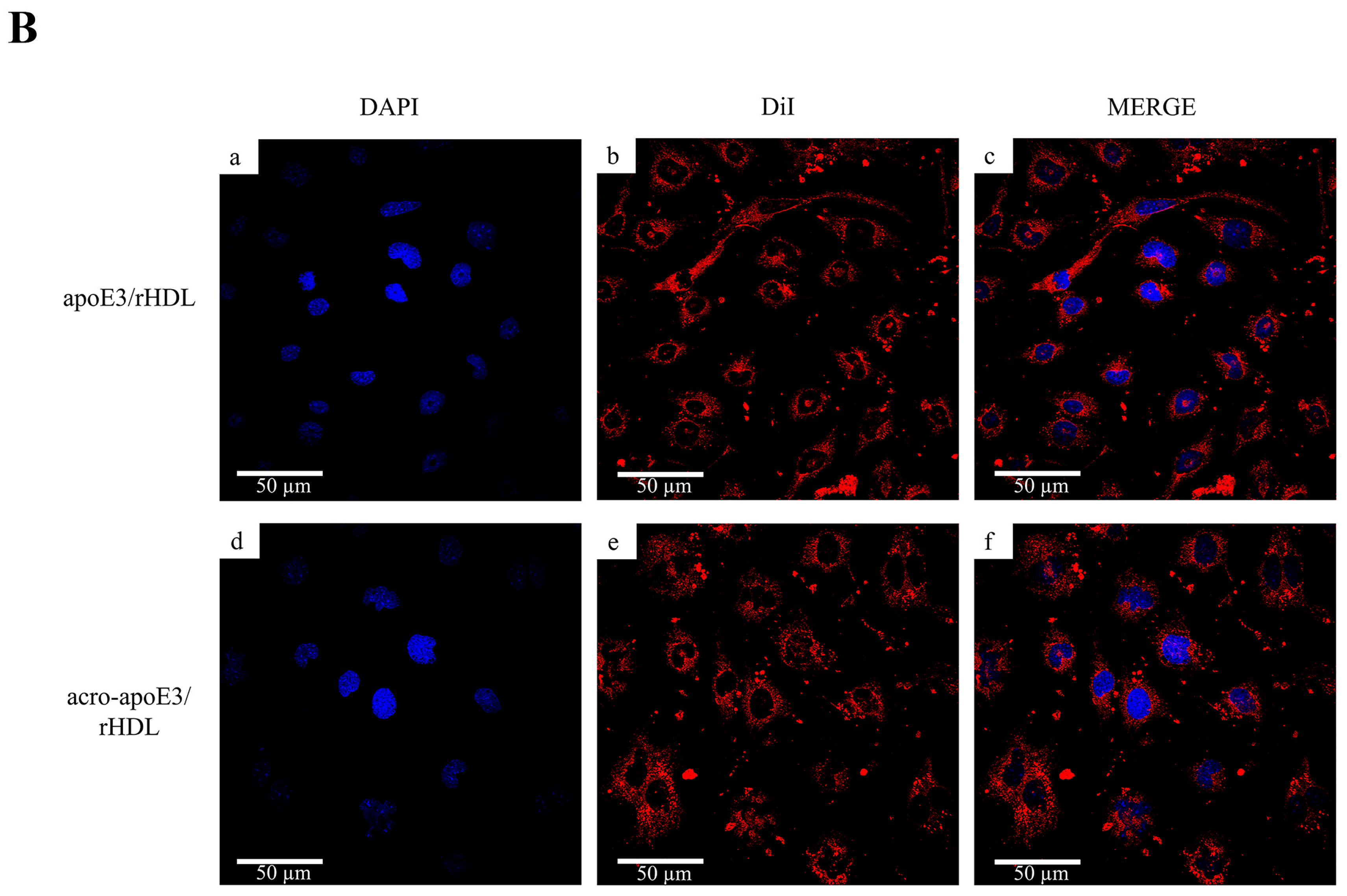
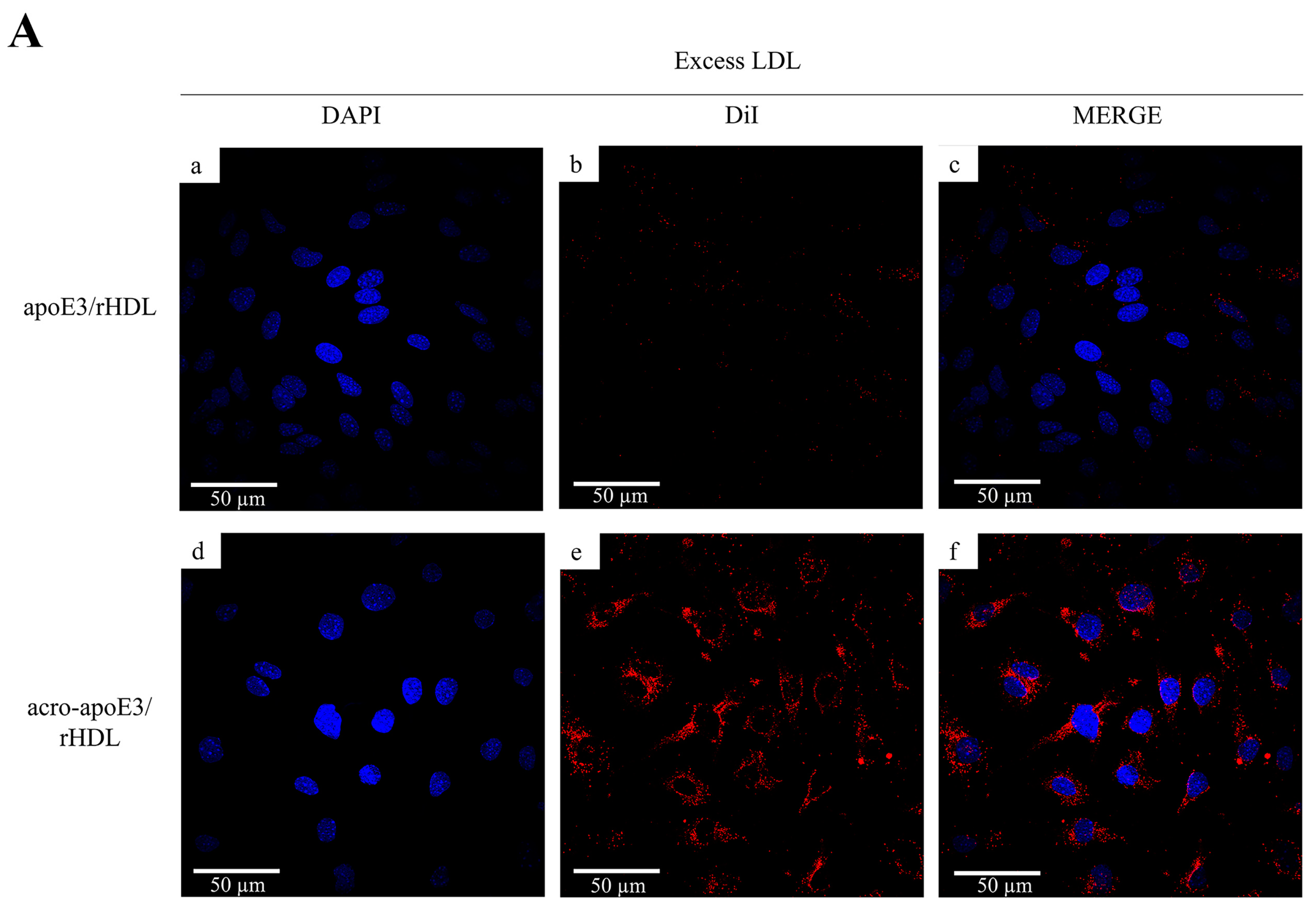
2.4. Cellular Uptake of acro-apoE3/rHDL
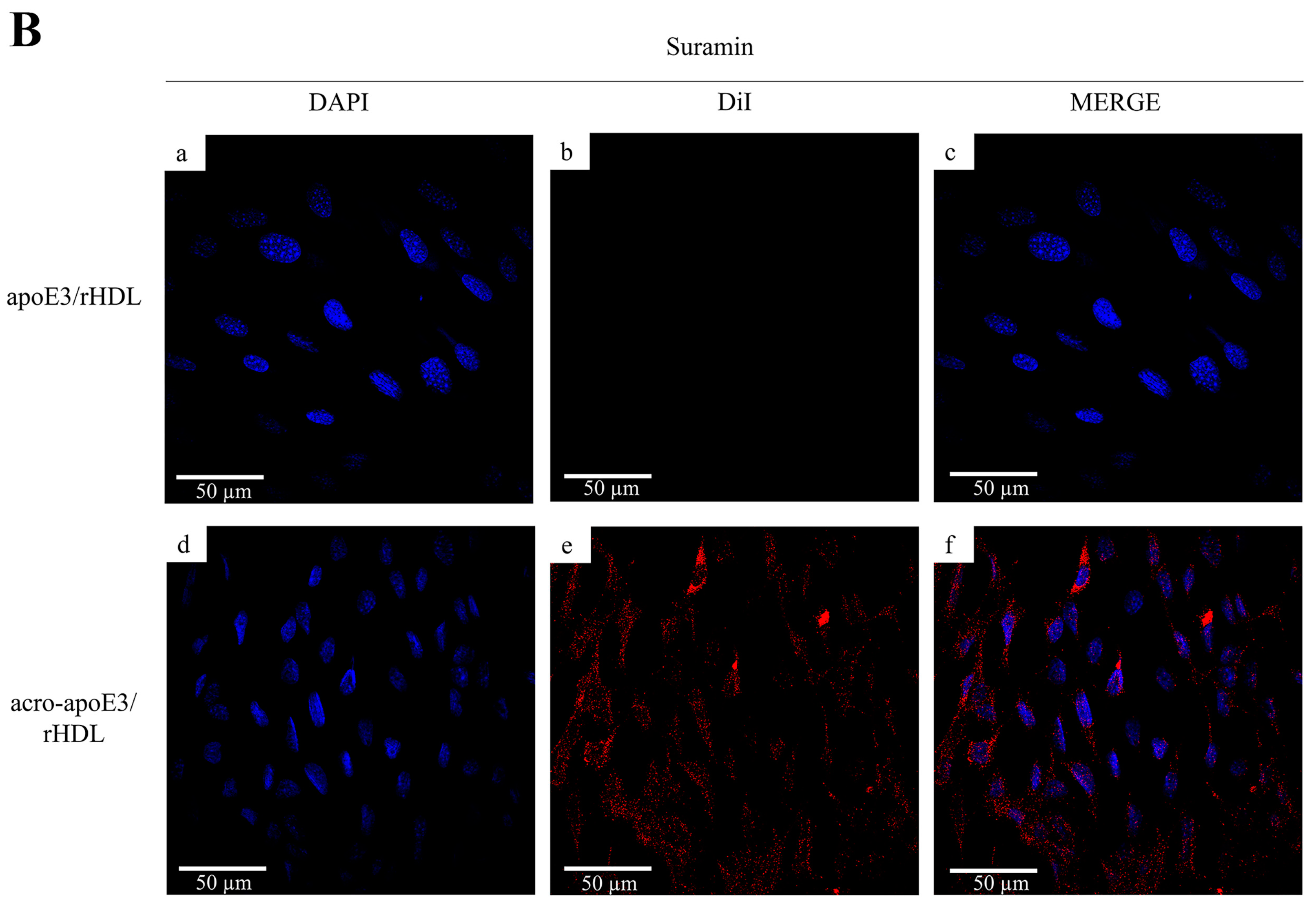
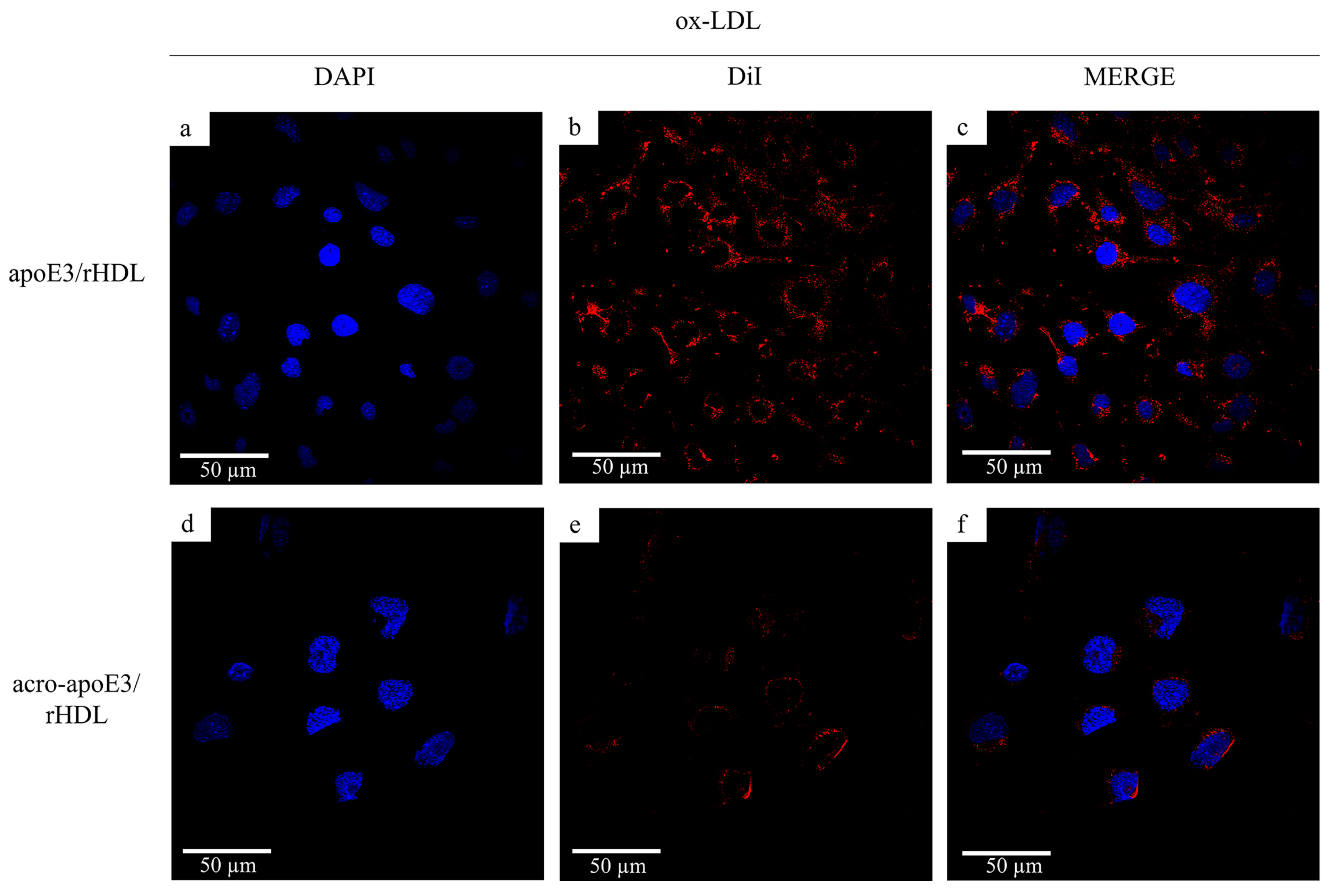
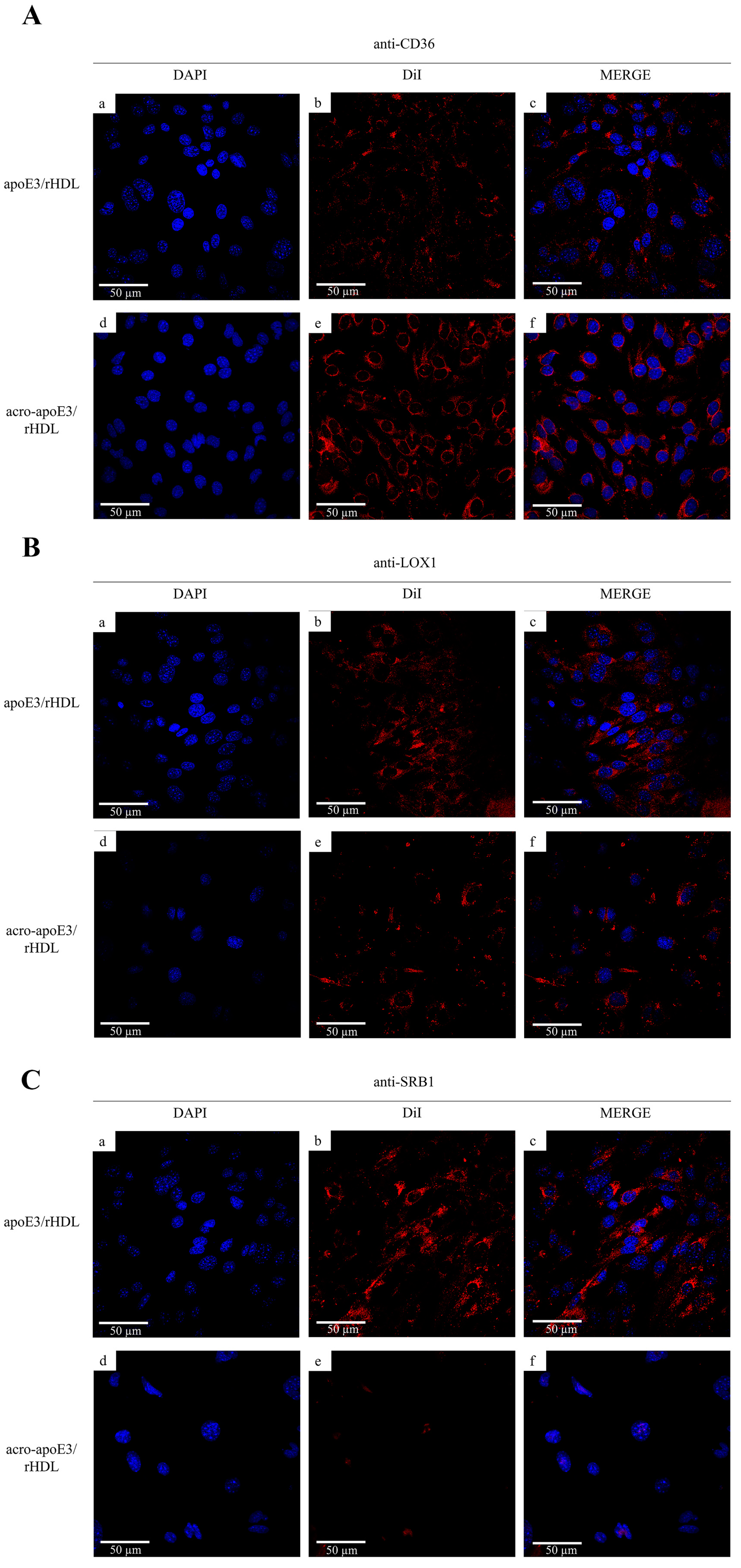
2.5. Internalization of Oxidatively Modified apoE3 by an Alternative Pathway
3. Discussion
3.1. Acrolein Forms Aldimine, Propanal, MP-Lys, and FDP-Lys Adducts in apoE3
3.2. Acro-apoE3/rHDL Does Not Interact with sLDLr and LDLr on bEnd.3 Cells
3.3. Acrolein-Modified apoE3 Is Internalized by LOX1 and SRB1 on bEnd.3 Cells
4. Materials and Methods
4.1. Acrolein Modification of apoE3
4.2. Western Blot
4.3. Circular Dichroism Spectroscopy
4.4. Mass Spectrometric Analysis
4.5. Preparation of rHDL
4.6. Non-Denaturing PAGE
4.7. Co-Immunoprecipitation
4.8. DiI Labeling of rHDL
4.9. Cellular Uptake of ApoE3/rHDL and Acro-apoE3/rHDL
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Mengi, S.A.; Xu, Y.-J.; Arneja, A.S.; Dhalla, N.S. Pathogenesis of atherosclerosis: A multifactorial process. Exp. Clin. Cardiol. 2002, 7, 40–53. [Google Scholar] [PubMed]
- Uno, M.; Kitazato, K.T.; Nishi, K.; Itabe, H.; Nagahiro, S. Raised plasma oxidised LDL in acute cerebral infarction. J. Neurol. Neurosurg. Psychiatry 2003, 74, 312. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.; Bacchetti, T.; Nègre-Salvayre, A.; Salvayre, R.; Dousset, N.; Curatola, G. Structural modifications of HDL and functional consequences. Atherosclerosis 2006, 184, 1–7. [Google Scholar] [CrossRef]
- Annema, W.; von Eckardstein, A. High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ. J. 2013, 77, 2432–2448. [Google Scholar] [CrossRef]
- Camont, L.; Chapman, M.J.; Kontush, A. Biological activities of HDL subpopulations and their relevance to cardiovascular disease. Trends Mol. Med. 2011, 17, 594–603. [Google Scholar] [CrossRef]
- Eren, E.; Yilmaz, N.; Aydin, O. High density lipoprotein and it’s dysfunction. Open Biochem. J. 2012, 6, 78–93. [Google Scholar] [CrossRef]
- Kontush, A.; Chapman, M.J. Functionally defective high-density lipoprotein: A new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol. Rev. 2006, 58, 342–374. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.M.; Mezdour, H.; Aratani, Y.; Knouff, C.; Najib, J.; Reddick, R.L.; Quarfordt, S.H.; Maeda, N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 1997, 272, 17972–17980. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.C. Apolipoprotein E isoforms and lipoprotein metabolism: Apolipoprotein E isoforms and lipoprotein metabolism. IUBMB Life 2014, 66, 616–623. [Google Scholar] [CrossRef] [PubMed]
- De Chaves, E.P.; Narayanaswami, V. Apolipoprotein E and cholesterol in aging and disease in the brain. Future Lipidol. 2008, 3, 505–530. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, R.; Mu, H.; Wang, X.; Yao, Q.; Chen, C. Reverse cholesterol transport and cholesterol efflux in atherosclerosis. QJM 2005, 98, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Jofre-Monseny, L.; Minihane, A.-M.; Rimbach, G. Impact of apoE genotype on oxidative stress, inflammation and disease risk. Mol. Nutr. Food Res. 2008, 52, 131–145. [Google Scholar] [CrossRef]
- Nishitsuji, K.; Hosono, T.; Nakamura, T.; Bu, G.; Michikawa, M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 2011, 286, 17536–17542. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Mahley, R.W. Apolipoprotein E: From cardiovascular disease to neurodegenerative disorders. J. Mol. Med. 2016, 94, 739–746. [Google Scholar] [CrossRef]
- Mahley, R.W.; Rall, S.C. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genomics. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.M.; Wilson, C.; Wardell, M.R.; Simmons, T.; Mahley, R.W.; Weisgraber, K.H.; Agard, D.A. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J. Biol. Chem. 1994, 269, 22358–22365. [Google Scholar] [PubMed]
- Faroon, O.; Roney, N.; Taylor, J.; Ashizawa, A.; Lumpkin, M.; Plewak, D. Acrolein environmental levels and potential for human exposure. Toxicol. Ind. Health 2008, 24, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kothari, S.; Patel, A.B.; Bielicki, J.K.; Narayanaswami, V. A pyrene based fluorescence approach to study conformation of apolipoprotein E3 in macrophage-generated nascent high density lipoprotein. Biochem. Biophys. Res. Commun. 2014, 450, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Tsujimoto, M. Endothelial scavenger receptors. Prog. Lipid Res. 2006, 45, 379–404. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Invest. 2001, 108, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Boullier, A.; Bird, D.A.; Chang, M.-K.; Dennis, E.A.; Friedman, P.; Gillotte-Taylor, K.; HöRkkö, S.; Palinski, W.; Quehenberger, O.; Shaw, P.; et al. Scavenger receptors, oxidized LDL, and atherosclerosis. Ann. N. Y. Acad. Sci. 2006, 947, 214–223. [Google Scholar] [CrossRef]
- Gillotte-Taylor, K.; Boullier, A.; Witztum, J.L.; Steinberg, D.; Quehenberger, O. Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. J. Lipid Res. 2001, 42, 1474–14782. [Google Scholar]
- Thorne, R.F.; Mhaidat, N.M.; Ralston, K.J.; Burns, G.F. CD36 is a receptor for oxidized high density lipoprotein: Implications for the development of atherosclerosis. FEBS Lett. 2007, 581, 1227–1232. [Google Scholar] [CrossRef]
- Chadwick, A.C.; Holme, R.L.; Chen, Y.; Thomas, M.J.; Sorci-Thomas, M.G.; Silverstein, R.L.; Pritchard, K.A.; Sahoo, D. Acrolein impairs the cholesterol transport functions of high density lipoproteins. PLoS ONE 2015, 10, e0123138. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Gokulan, K.; Mehta, J.L.; Varughese, K.I. Structure-based design targeted at LOX-1, a receptor for oxidized low-density lipoprotein. Sci. Rep. 2015, 5, 16740. [Google Scholar] [CrossRef] [PubMed]
- Enciu, A.-M.; Gherghiceanu, M.; Popescu, B.O. Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxid. Med. Cell. Longev. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Yoshida, M.; Waragai, M.; Kashiwagi, K. Evaluation of dementia by acrolein, amyloid-β and creatinine. Clin. Chim. Acta 2015, 450, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Kalhorn, T.F.; Slattery, J.T. Inhibition of human aldehyde dehydrogenase 1 by the 4-hydroxycyclophosphamide degradation product acrolein. Drug Metab. Dispos. 1999, 27, 133–137. [Google Scholar] [PubMed]
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol. Aging 2001, 22, 187–194. [Google Scholar] [CrossRef]
- Sakata, K.; Kashiwagi, K.; Sharmin, S.; Ueda, S.; Irie, Y.; Murotani, N.; Igarashi, K. Increase in putrescine, amine oxidase, and acrolein in plasma of renal failure patients. Biochem. Biophys. Res. Commun. 2003, 305, 143–149. [Google Scholar] [CrossRef]
- Shao, B.; O’Brien, K.D.; Mcdonald, T.O.; Fu, X.; Oram, J.F.; Uchida, K.; Heinecke, J.W. Acrolein modifies apolipoprotein A-I in the human artery wall. Ann. N. Y. Acad. Sci. 2005, 1043, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Fu, X.; McDonald, T.O.; Green, P.S.; Uchida, K.; O’Brien, K.D.; Oram, J.F.; Heinecke, J.W. Acrolein impairs ATP binding cassette transporter A1-dependent cholesterol export from cells through site-specific modification of apolipoprotein A-I. J. Biol. Chem. 2005, 280, 36386–36396. [Google Scholar] [CrossRef]
- Uchida, K.; Kanematsu, M.; Morimitsu, Y.; Osawa, T.; Noguchi, N.; Niki, E. Acrolein is a product of lipid peroxidation reaction: formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. J. Biol. Chem. 1998, 273, 16058–16066. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K.; Kanematsu, M.; Sakai, K.; Matsuda, T.; Hattori, N.; Mizuno, Y.; Suzuki, D.; Miyata, T.; Noguchi, N.; Niki, E.; et al. Protein-bound acrolein: Potential markers for oxidative stress. Proc. Natl. Acad. Sci. USA 1998, 95, 4882–4887. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. Current status of acrolein as a lipid peroxidation product. Trends Cardiovasc. Med. 1999, 9, 109–113. [Google Scholar] [CrossRef]
- Furuhata, A.; Ishii, T.; Kumazawa, S.; Yamada, T.; Nakayama, T.; Uchida, K. N(epsilon)-(3-methylpyridinium)lysine, a major antigenic adduct generated in acrolein-modified protein. J. Biol. Chem. 2003, 278, 48658–48665. [Google Scholar] [CrossRef] [PubMed]
- Jairam, V.; Uchida, K.; Narayanaswami, V. Pathophysiology of Lipoprotein Oxidation. In Lipoproteins—Role in Health and Diseases; Frank, S., Kostner, G., Eds.; InTech: New York, NY, USA, 2012; pp. 383–408. [Google Scholar]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Lund-Katz, S.; Zaiou, M.; Wehrli, S.; Dhanasekaran, P.; Baldwin, F.; Weisgraber, K.H.; Phillips, M.C. Effects of lipid interaction on the lysine microenvironments in apolipoprotein E. J. Biol. Chem. 2000, 275, 34459–34464. [Google Scholar] [CrossRef] [PubMed]
- Lund-Katz, S.; Wehrli, S.; Zaiou, M.; Newhouse, Y.; Weisgraber, K.H.; Phillips, M.C. Effects of polymorphism on the microenvironment of the LDL receptor-binding region of human apoE. J. Lipid Res. 2001, 42, 894–901. [Google Scholar]
- Tran, T.N.; Kosaraju, M.G.; Tamamizu-Kato, S.; Akintunde, O.; Zheng, Y.; Bielicki, J.K.; Pinkerton, K.; Uchida, K.; Lee, Y.Y.; Narayanaswami, V. Acrolein modification impairs key functional features of rat apolipoprotein E: Identification of modified sites by mass spectrometry. Biochemistry 2014, 53, 361–375. [Google Scholar] [CrossRef]
- Raussens, V.; Fisher, C.A.; Goormaghtigh, E.; Ryan, R.O.; Ruysschaert, J.-M. The low density lipoprotein receptor active conformation of apolipoprotein E: helix organization in n-terminal domain-phospholipid disc particles. J. Biol. Chem. 1998, 273, 25825–25830. [Google Scholar] [CrossRef][Green Version]
- Dhaliwal, B.S.; Steinbrecher, U.P. Scavenger receptors and oxidized low density lipoproteins. Clin. Chim. Acta 1999, 286, 191–205. [Google Scholar] [CrossRef]
- Yoshida, H.; Quehenberger, O.; Kondratenko, N.; Green, S.; Steinberg, D. Minimally oxidized low-density lipoprotein increases expression of scavenger receptor A, CD36, and macrosialin in resident mouse peritoneal macrophages. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Levitan, I.; Volkov, S.; Subbaiah, P.V. Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox. Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef] [PubMed]
- Boullier, A.; Gillotte, K.L.; Hörkkö, S.; Green, S.R.; Friedman, P.; Dennis, E.A.; Witztum, J.L.; Steinberg, D.; Quehenberger, O. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J. Biol. Chem. 2000, 275, 9163–9169. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Sawamura, T.; Wakabayashi, I.; Okamura, T. LOX-1 in atherosclerotic disease. Clin. Chim. Acta 2015, 440, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J. Clin. Investig. 2001, 108, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Krieger, M. Charting the fate of the “good cholesterol”: Identification and characterization of the high-density lipoprotein receptor SR-BI. Annu. Rev. Biochem. 1999, 68, 523–558. [Google Scholar] [CrossRef]
- Morrow, J.A.; Segall, M.L.; Lund-Katz, S.; Phillips, M.C.; Knapp, M.; Rupp, B.; Weisgraber, K.H. Differences in stability among the human apolipoprotein E isoforms determined by the amino-terminal domain. Biochemistry 2000, 39, 11657–11666. [Google Scholar] [CrossRef]
- Kim, S.H.; Adhikari, B.B.; Cruz, S.; Schramm, M.P.; Vinson, J.A.; Narayanaswami, V. Targeted intracellular delivery of resveratrol to glioblastoma cells using apolipoprotein E-containing reconstituted HDL as a nanovehicle. PLoS ONE 2015, 10, e0135130. [Google Scholar] [CrossRef]
- Steinbrecher, U.P. Oxidation of human low density lipoprotein results in derivatization of lysine residues of apolipoprotein B by lipid peroxide decomposition products. J. Biol. Chem. 1987, 262, 3603–3608. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trypsin | Elastase | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Site | Mass Increase | † Best ASCORE | Spectral Count Acro-ApoE3 | Nature of Lys Modification | Site | Mass Increase | † Best Ascore | Spectral Count Acro-ApoE3 | Nature of Modification |
| K1 | - | - | - | - | K1 | 76 | 1000 | 3 | MP-Lys |
| K69 | 76 | 161.94 | 9 | MP-Lys § | K69 | 76 | 1000 | 12 | MP-Lys |
| K69 | 56 | 100.09 | 3 | Propanal adduct | K69 | 56 | 100.09 | 2 | Propanal adduct |
| K69 | 38 | 1000 | 1 | Aldimine | K69 | - | - | - | - |
| K72 | 76 | 119.87 | 9 | MP-Lys | K72 | 76 | 153.17 | 15 | MP-Lys |
| K72 | 38 | 1000 | 1 | Aldimine | K72 | 38 | 1000 | 2 | Aldimine |
| K75 | 76 | 1000 | 6 | MP-Lys | K75 | 76 | 1000 | 8 | MP-Lys |
| K75 | 94 | 1000 | 1 | FDP-Lys †† | K75 | 94 | 1000 | 1 | FDP-Lys |
| K75 | 56 | 120.17 | 2 | Propanal adduct | K75 | - | - | - | - |
| K75 | 38 | 1000 | 1 | Aldimine | K75 | - | - | - | - |
| K95 | 76 | 1000 | 5 | MP-Lys | K95 | 76 | 1000 | 4 | MP-Lys |
| K95 | 94 | 1000 | 1 | FDP-Lys | K95 | 94 | 1000 | 1 | FDP-Lys |
| K143 | - | - | - | - | - | - | - | - | - |
| K146 | - | - | - | - | - | - | - | - | - |
| K157 | 76 | 1000 | 6 | MP-Lys | K157 | 76 | 1000 | 9 | MP-Lys |
| K233 | 76 | 1000 | 9 | MP-Lys | K233 | 76 | 1000 | 11 | MP-Lys |
| K233 | 56 | 1000 | 4 | Propanal adduct | K233 | 56 | 1000 | 2 | Propanal adduct |
| K233 | 38 | 1000 | 1 | Aldimine | K233 | 38 | 1000 | 1 | Aldimine |
| K242 | 76 | 1000 | 4 | MP-Lys | K242 | 76 | 1000 | 4 | MP-Lys |
| K242 | 94 | 1000 | 2 | FDP-Lys | K242 | 94 | 1000 | 2 | FDP-Lys |
| K262 | 76 | 1000 | 6 | MP-Lys | K262 | 76 | 1000 | 8 | MP-Lys |
| K282 | 76 | 1000 | 1 | MP-Lys | K282 | 76 | 1000 | 3 | MP-Lys |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, S.; Narayanaswami, V. Cellular Uptake and Clearance of Oxidatively-modified Apolipoprotein E3 by Cerebral Cortex Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 4582. https://doi.org/10.3390/ijms20184582
Cruz S, Narayanaswami V. Cellular Uptake and Clearance of Oxidatively-modified Apolipoprotein E3 by Cerebral Cortex Endothelial Cells. International Journal of Molecular Sciences. 2019; 20(18):4582. https://doi.org/10.3390/ijms20184582
Chicago/Turabian StyleCruz, Siobanth, and Vasanthy Narayanaswami. 2019. "Cellular Uptake and Clearance of Oxidatively-modified Apolipoprotein E3 by Cerebral Cortex Endothelial Cells" International Journal of Molecular Sciences 20, no. 18: 4582. https://doi.org/10.3390/ijms20184582
APA StyleCruz, S., & Narayanaswami, V. (2019). Cellular Uptake and Clearance of Oxidatively-modified Apolipoprotein E3 by Cerebral Cortex Endothelial Cells. International Journal of Molecular Sciences, 20(18), 4582. https://doi.org/10.3390/ijms20184582