Chirality-Dependent Adsorption between Amphipathic Peptide and POPC Membrane


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Why Adsorption of the Peptide Is Interested
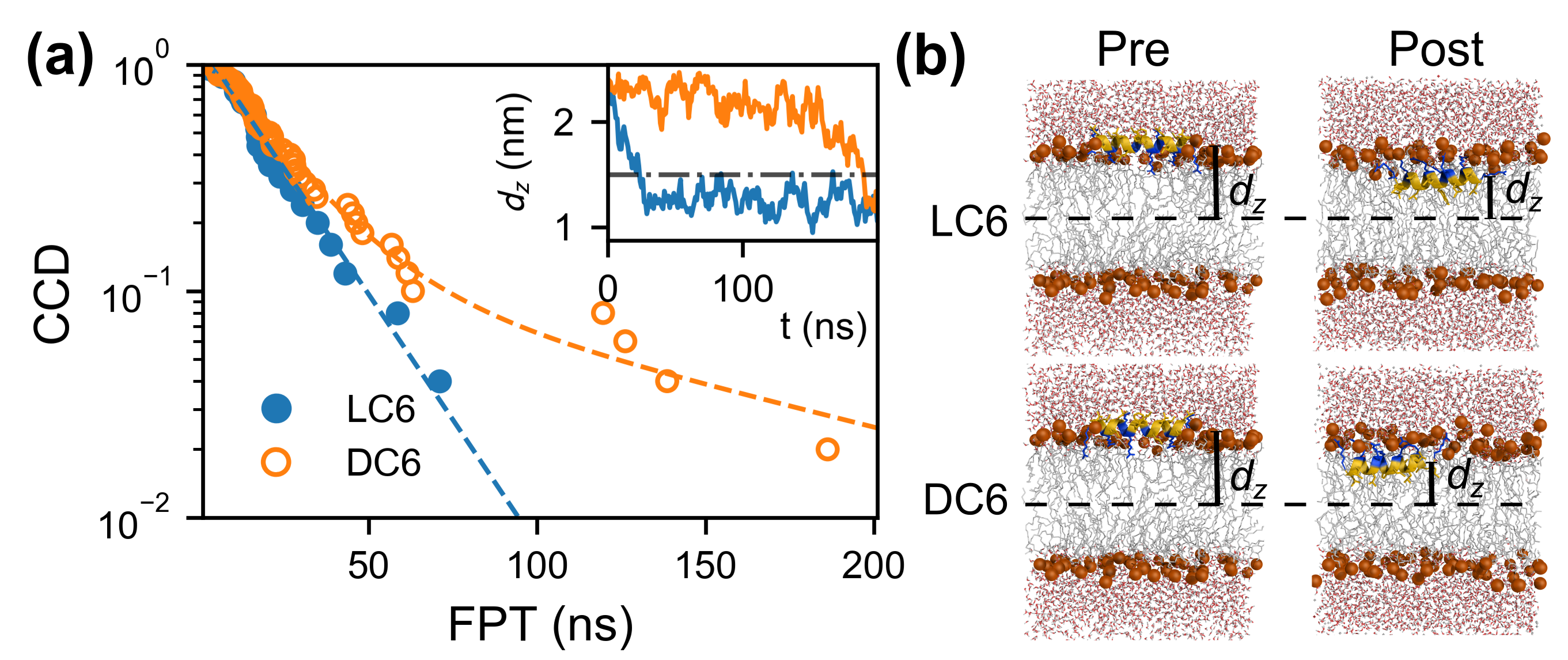
2.2. Adsorption of Monomeric Peptide C6
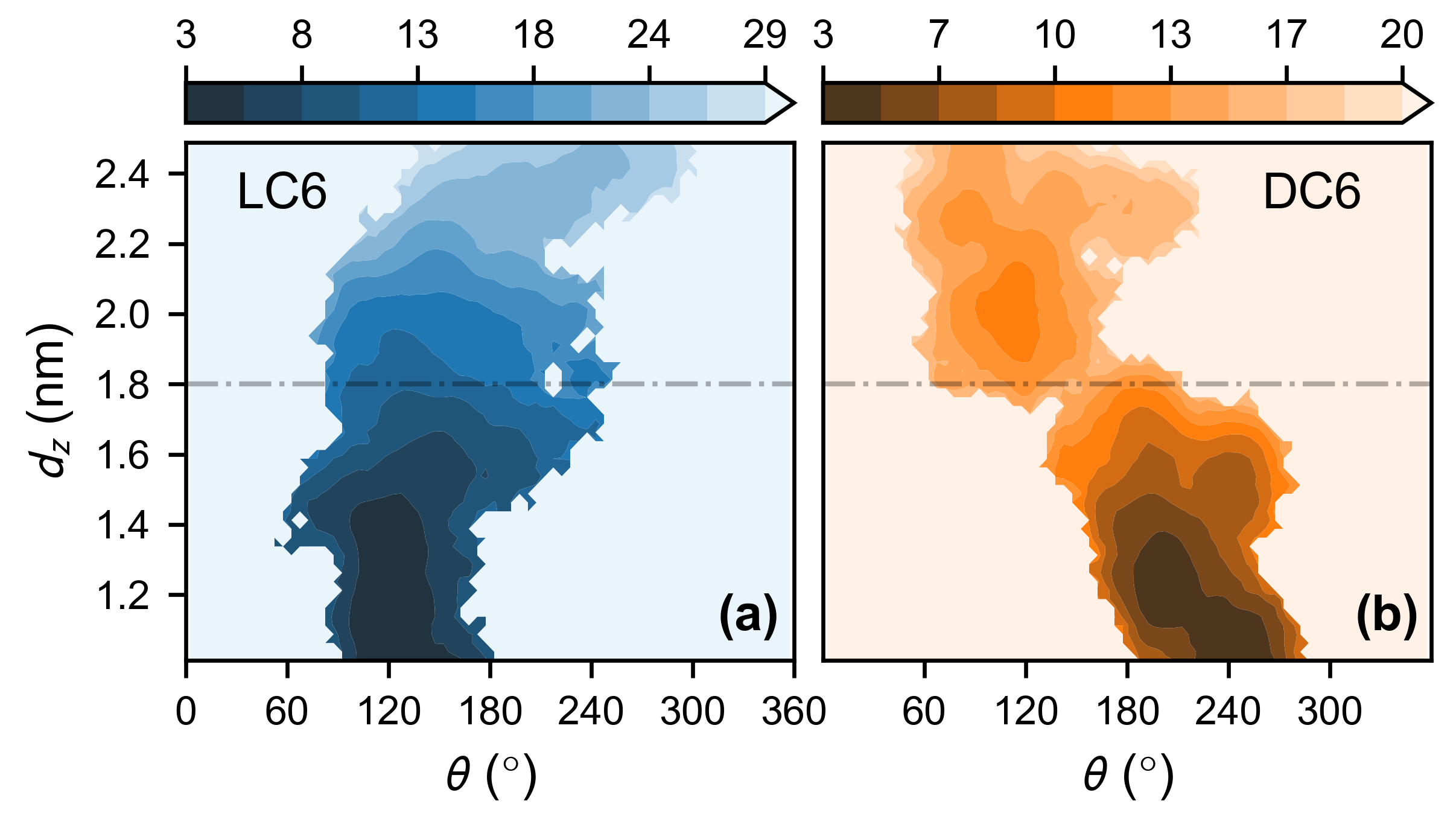
2.3. Umbrella Sampling on Typical Pathways
2.4. Differences in Contact Interactions During the Adsorption of C6 Enantiomers
2.5. Nonuniform Distribution of Lipids around the Peptide
2.6. The Key Role of Trp
3. Materials and Methods
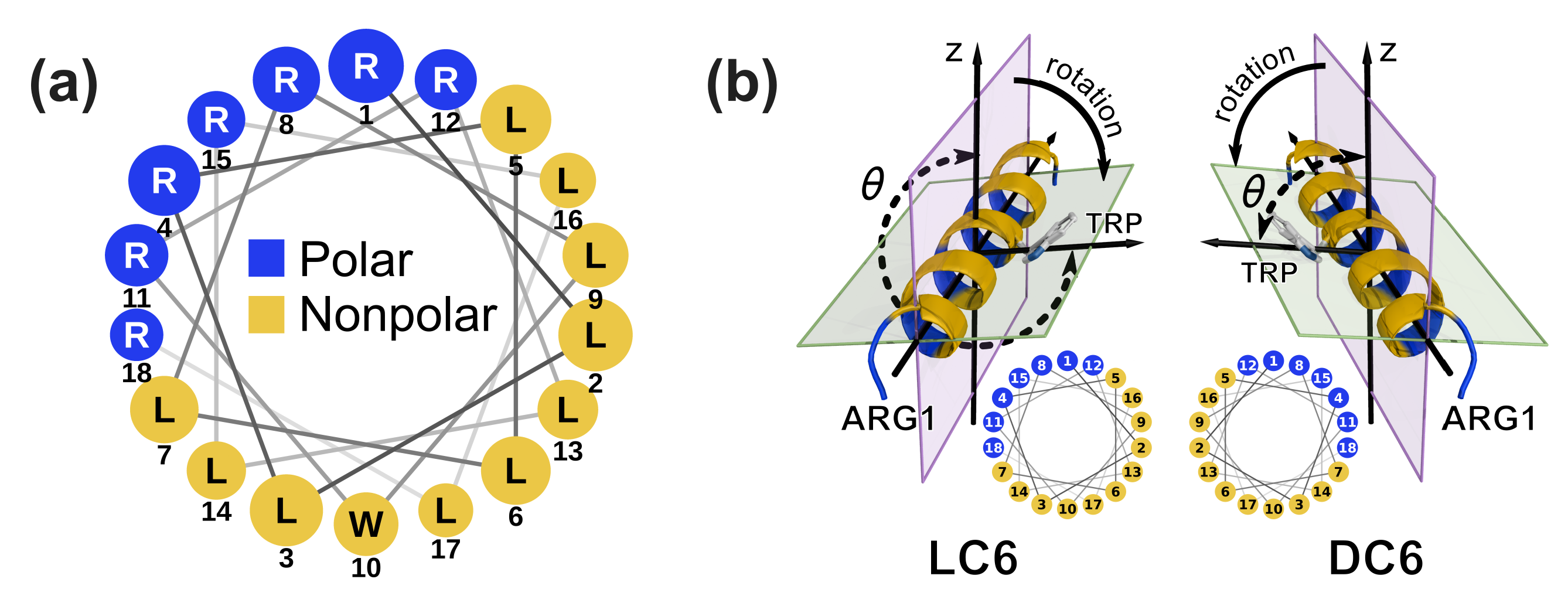
3.1. System Preparation
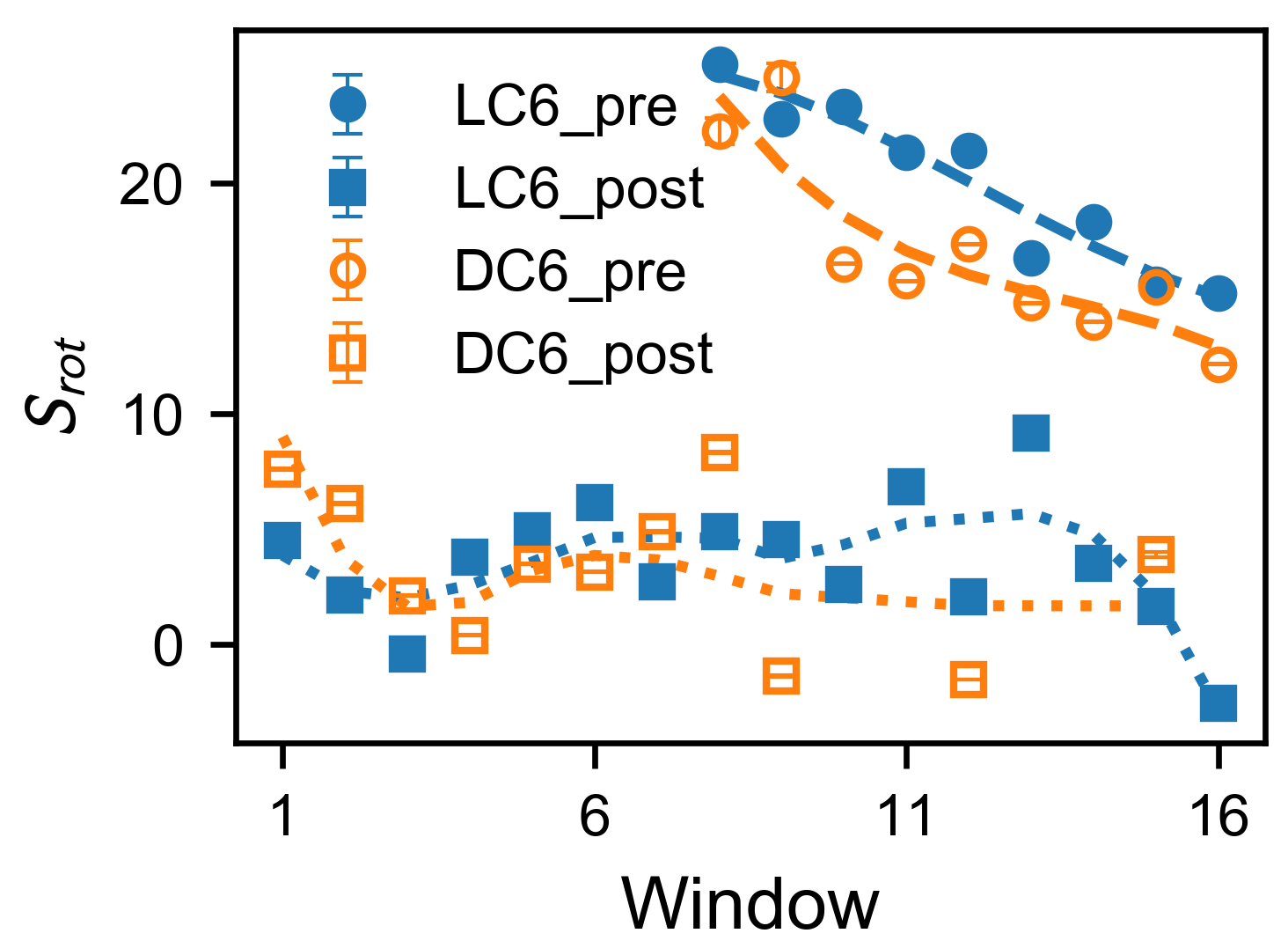
3.2. Characterization of Helix Rotation
3.3. Adsorption of Monomeric Peptide
3.4. Umbrella Sampling for the Adsorption Free Energy
3.5. Analysis on Umbrella Windows
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | antimicrobial peptide |
| LC6 | the C6 peptide composed of L-type amino acids |
| DC6 | the C6 peptide composed of D-type amino acids |
| COM | center of mass |
| CCD | complementary cumulative distribution |
| FPT | first passage time |
| PMF | potential of mean force |
| CV | collective variable |
| SEM | standard error of the mean |
| MBAR | multistate Bennett acceptance ratio estimator |
References
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutti, F.G.; Knaus, T.; Scrutton, N.S.; Breuer, M.; Turner, N.J. Conversion of Alcohols to Enantiopure Amines Through Dual-Enzyme Hydrogen-Borrowing Cascades. Science 2015, 349, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.; Boman, A.; Wåhlin, B.; Drain, C.M.; Andreu, D.; Boman, H.G.; Merrifield, R.B. All-D Amino Acid-Containing Channel-Forming Antibiotic Peptides. Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Arribart, H.; Guille, M.M.G. Biomimetism and Bioinspiration as Tools for the Design of Innovative Materials and Systems. Nat. Mater. 2005, 4, 277. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.X.; Cross, T.A. Modeling the Membrane Environment Has Implications for Membrane Protein Structure and Function: Influenza a M2 Protein. Protein Sci. 2013, 22, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Baulin, V.A.; Thalmann, F. Peroxidised Phospholipid Bilayers: Insight from Coarse-Grained Molecular Dynamics Simulations. Soft Matter 2015, 12, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Baulin, V. GPU Implementation of the Rosenbluth Generation Method for Static Monte Carlo Simulations. Comput. Phys. Commun. 2017, 216, 95–101. [Google Scholar] [CrossRef]
- Ramadurai, S.; Werner, M.; Slater, N.K.H.; Martin, A.; Baulin, V.A.; Keyes, T.E. Dynamic studies of the interaction of a pH responsive, amphiphilic polymer with a DOPC lipid membrane. Soft Matter 2017, 13, 3690–3700. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.M.; Yuan, B.; Zhang, X.R.; Yang, K.; Ma, Y.Q. Molecular Modeling of Transmembrane Delivery of Paclitaxel by Shock Waves with Nanobubbles. Appl. Phys. Lett. 2017, 110, 023701. [Google Scholar] [CrossRef]
- Setzler, J.; Seith, C.; Brieg, M.; Wenzel, W. SLIM: An Improved Generalized Born Implicit Membrane Model. J. Comput. Chem. 2014, 35, 2027–2039. [Google Scholar] [CrossRef]
- Feig, M. Implicit Membrane Models for Membrane Protein Simulation. In Methods in Molecular Biology; Kukol, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; Volume 443, pp. 181–196. [Google Scholar]
- Yuzlenko, O.; Lazaridis, T. Membrane Protein Native State Discrimination by Implicit Membrane Models. J. Comput. Chem. 2013, 34, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Ulmschneider, J.P.; Ulmschneider, M.B. Sampling Efficiency in Explicit and Implicit Membrane Environments Studied by Peptide Folding Simulations. Proteins Struct. Funct. Bioinf. 2009, 75, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Arranz-Gibert, P.; Guixer, B.; Malakoutikhah, M.; Muttenthaler, M.; Guzmán, F.; Teixidó, M.; Giralt, E. Lipid Bilayer Crossing—The Gate of Symmetry. Water-Soluble Phenylproline-Based Blood-Brain Barrier Shuttles. J. Am. Chem. Soc. 2015, 137, 7357–7364. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kishi, Y.; Ishigami, T.; Suga, K.; Umakoshi, H. Chiral Selective Adsorption of Ibuprofen on a Liposome Membrane. J. Phys. Chem. B 2016, 120, 2790–2795. [Google Scholar] [CrossRef]
- Novotná, P.; Goncharova, I.; Urbanová, M. Mutual Structural Effect of Bilirubin and Model Membranes by Vibrational Circular Dichroism. Biochim. Biophys. Acta Biomembr. 2014, 1838, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Novotná, P.; Králík, F.; Urbanová, M. Chiral Recognition of Bilirubin and Biliverdin in Liposomes and Micelles. Biophys. Chem. 2015, 205, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, T.; Suga, K.; Umakoshi, H. Chiral Recognition of L-Amino Acids on Liposomes Prepared with L-Phospholipid. ACS Appl. Mater. Interfaces 2015, 7, 21065–21072. [Google Scholar] [CrossRef]
- Sarangi, N.K.; Ramesh, N.; Patnaik, A. Structure and Dynamics of H2O Vis-Á-Vis Phenylalanine Recognition at a DPPC Lipid Membrane Via Interfacial H-Bond Types: Insights from Polarized FT-IRRAS and ADMP Simulations. J. Chem. Phys. 2015, 142, 024702. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, Z.; Qing, G. Interfacial Interaction on Phospholipid Membrane. Prog. Chem. 2018, 30, 888–901. [Google Scholar] [CrossRef]
- Bombelli, C.; Borocci, S.; Lupi, F.; Mancini, G.; Mannina, L.; Segre, A.L.; Viel, S. Chiral Recognition of Dipeptides in a Biomembrane Model. J. Am. Chem. Soc. 2004, 126, 13354–13362. [Google Scholar] [CrossRef]
- Bombelli, C.; Borocci, S.; Cruciani, O.; Mancini, G.; Monti, D.; Segre, A.L.; Sorrenti, A.; Venanzi, M. Chiral Recognition of Dipeptides in Bio-Membrane Models: The Role of Amphiphile Hydrophobic Chains. Tetrahedron Asymmetry 2008, 19, 124–130. [Google Scholar] [CrossRef]
- Cruciani, O.; Borocci, S.; Lamanna, R.; Mancini, G.; Segre, A.L. Chiral Recognition of Dipeptides in Phosphatidylcholine Aggregates. Tetrahedron Asymmetry 2006, 17, 2731–2737. [Google Scholar] [CrossRef]
- Lopes, S.C.D.N.; Fedorov, A.; Castanho, M.A.R.B. Chiral Recognition of D-Kyotorphin by Lipidic Membranes: Relevance Toward Improved Analgesic Efficiency. ChemMedChem 2006, 1, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, A.; Diociaiuti, M.; Corvaglia, V.; Chistolini, P.; Mancini, G. Chiral Recognition of Dipeptides in Langmuir Monolayers. Tetrahedron Asymmetry 2009, 20, 2737–2741. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Mizogami, M. The Membrane Interaction of Drugs as One of Mechanisms for Their Enantioselective Effects. Med. Hypotheses 2012, 79, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Xu, W.; Naahidi, S.; Chen, B.; Chen, P. A New Amphipathic, Amino-Acid-Pairing (AAP) Peptide as siRNA Delivery Carrier: Physicochemical Characterization and in Vitro Uptake. J. Phys. Chem. B 2012, 116, 13183–13191. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, K.; Sheng, Y.; Wang, J.; Wang, W.; Chen, P. Chiral C6 Interact Differently with POPC Membrane. manuscript in preparation. 2019. [Google Scholar]
- Juba, M.; Porter, D.; Dean, S.; Gillmor, S.; Bishop, B. Characterization and Performance of Short Cationic Antimicrobial Peptide Isomers. Biopolymers 2013, 100, 387–401. [Google Scholar] [CrossRef]
- Sando, L.; Henriques, S.T.; Foley, F.; Simonsen, S.M.; Daly, N.L.; Hall, K.N.; Gustafson, K.R.; Aguilar, M.I.; Craik, D.J. A Synthetic Mirror Image of Kalata B1 Reveals that Cyclotide Activity Is Independent of a Protein Receptor. ChemBioChem 2011, 12, 2456–2462. [Google Scholar] [CrossRef]
- Henriques, S.T.; Craik, D.J. Importance of the Cell Membrane on the Mechanism of Action of Cyclotides. ACS Chem. Biol. 2012, 7, 626–636. [Google Scholar] [CrossRef]
- Ulmschneider, J.P.; Ulmschneider, M.B. Molecular Dynamics Simulations Are Redefining Our View of Peptides Interacting with Biological Membranes. Acc. Chem. Res. 2018, 51, 1106–1116. [Google Scholar] [CrossRef]
- Hong, S.Y.; Oh, J.E.; Lee, K.H. Effect of D-amino acid substitution on the stability, the secondary structure, and the activity of membrane-active peptide. Biochem. Pharmacol. 1999, 58, 1775–1780. [Google Scholar] [CrossRef]
- Krugliak, M.; Feder, R.; Zolotarev, V.Y.; Gaidukov, L.; Dagan, A.; Ginsburg, H.; Mor, A. Antimalarial Activities of Dermaseptin S4 Derivatives. Antimicrob. Agents Chemother. 2000, 44, 2442–2451. [Google Scholar] [CrossRef] [Green Version]
- Wade, D.; Silberring, J.; Soliymani, R.; Heikkinen, S.; Kilpelainen, I.; Lankinen, H.; Kuusela, P. Antibacterial Activities of Temporin A Analogs. FEBS Lett. 2000, 479, 6–9. [Google Scholar] [CrossRef]
- Hauge, H.H.; Mantzilas, D.; Moll, G.N.; Konings, W.N.; Driessen, A.J.M.; Eijsink, V.G.H.; Nissen-Meyer, J. Plantaricin A Is an Amphiphilic Alpha-Helical Bacteriocin-Like Pheromone Which Exerts Antimicrobial and Pheromone Activities Through Different Mechanisms. Biochemistry 1998, 37, 16026–16032. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, P.E.; Fimland, G.; Mantzilas, D.; Nissen-Meyer, J. Structure and Mode of Action of the Membrane-Permeabilizing Antimicrobial Peptide Pheromone Plantaricin A. J. Biol. Chem. 2005, 280, 22945–22950. [Google Scholar] [CrossRef] [PubMed]
- Providence, L.L.; Andersen, O.S.; Greathouse, D.V.; Koeppe, R.E.; Bittman, R. Gramicidin channel function does not depend on phospholipid chirality. Biochemistry 1995, 34, 16404–16411. [Google Scholar] [CrossRef]
- Wang, C.K.; King, G.J.; Conibear, A.C.; Ramos, M.C.; Chaousis, S.; Henriques, S.T.; Craik, D.J. Mirror Images of Antimicrobial Peptides Provide Reflections on Their Functions and Amyloidogenic Properties. J. Am. Chem. Soc. 2016. [Google Scholar] [CrossRef]
- Wei, G.; Leeuw, E.D.; Pazgier, M.; Yuan, W.; Zou, G.; Wang, J.; Ericksen, B.; Lu, W.Y.; Lehrer, R.I.; Lu, W. Through the Looking Glass, Mechanistic Insights from Enantiomeric Human Defensins. J. Biol. Chem. 2009, 284, 29180–29192. [Google Scholar] [CrossRef] [Green Version]
- Merrifield, E.L.; Mitchell, S.A.; Ubach, J.; Boman, H.G.; Andreu, D.; Merrifield, R.B. D-Enantiomers of 15-Residue Cecropin A-Melittin Hybrids. Int. J. Pept. Protein Res. 1995, 46, 214–220. [Google Scholar] [CrossRef]
- Huang, J.; Hao, D.; Chen, Y.; Xu, Y.; Tan, J.; Huang, Y.; Li, F.; Chen, Y. Inhibitory Effects and Mechanisms of Physiological Conditions on the Activity of Enantiomeric Forms of an α-Helical Antibacterial Peptide Against Bacteria. Peptides 2011, 32, 1488–1495. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yount, N.Y.; Bayer, A.S.; Xiong, Y.Q.; Yeaman, M.R. Advances in antimicrobial peptide immunobiology. Biopolymers 2006, 84, 435–458. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial Peptides in Action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal Pores Formed by Antimicrobial Peptides Show Significant Disorder. Biochim. Biophys. Acta Biomembr. 2008, 1778, 2308–2317. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, S.A.; Böckmann, R.A. Membrane Pore Formation in Atomistic and Coarse-Grained Simulations. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2266–2277. [Google Scholar] [CrossRef]
- Ludtke, S.J.; He, K.; Heller, W.T.; Harroun, T.A.; Yang, L.; Huang, H.W. Membrane Pores Induced by Magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, T.; Wei, D.; Strandberg, E.; Ulrich, A.S.; Ulmschneider, J.P. How Reliable Are Molecular Dynamics Simulations of Membrane Active Antimicrobial Peptides? Biochim. Biophys. Acta Biomembr. 2014, 1838, 2280–2288. [Google Scholar] [CrossRef]
- Irudayam, S.J.; Berkowitz, M.L. Binding and Reorientation of Melittin in a POPC Bilayer: Computer Simulations. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2975–2981. [Google Scholar] [CrossRef]
- Mol, A.R.; Castro, M.S.; Fontes, W. Netwheels: A Web Application to Create High Quality Peptide Helical Wheel and Net Projections. bioRxiv 2018, 416347. [Google Scholar] [CrossRef]
- Sun, D.; Forsman, J.; Woodward, C.E. Multistep Molecular Dynamics Simulations Identify the Highly Cooperative Activity of Melittin in Recognizing and Stabilizing Membrane Pores. Langmuir 2015, 31, 9388–9401. [Google Scholar] [CrossRef] [Green Version]
- Esbjörner, E.K.; Caesar, C.E.B.; Albinsson, B.; Lincoln, P.; Nordén, B. Tryptophan Orientation in Model Lipid Membranes. Biochem. Biophys. Res. Commun. 2007, 361, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Gaede, H.C.; Yau, W.M.; Gawrisch, K. Electrostatic Contributions to Indole-Lipid Interactions. J. Phys. Chem. B 2005, 109, 13014–13023. [Google Scholar] [CrossRef] [PubMed]
- Persson, S.; Killian, J.A.; Lindblom, G. Molecular Ordering of Interfacially Localized Tryptophan Analogs in Ester- and Ether-Lipid Bilayers Studied by 2H-NMR. Biophys. J. 1998, 75, 1365–1371. [Google Scholar] [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.2; Schrödinger, LLC: Cambridge, MA, USA, 2018.
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed]
- Pluhackova, K.; Kirsch, S.A.; Han, J.; Sun, L.; Jiang, Z.; Unruh, T.; Böckmann, R.A. A Critical Comparison of Biomembrane Force Fields: Structure and Dynamics of Model DMPC, POPC, and POPE Bilayers. J. Phys. Chem. B 2016, 120, 3888–3903. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Ulmschneider, J.; Smith, J.; Ulmschneider, M.; Ulrich, A.; Strandberg, E. Reorientation and Dimerization of the Membrane-Bound Antimicrobial Peptide PGLa from Microsecond All-Atom MD Simulations. Biophys. J. 2012, 103, 472–482. [Google Scholar] [CrossRef] [Green Version]
- Ulmschneider, M.B.; Sansom, M.S.P.; Di Nola, A. Evaluating Tilt Angles of Membrane-Associated Helices: Comparison of Computational and NMR Techniques. Biophys. J. 2006, 90, 1650–1660. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, C.H.; Hu, D.; Ulmschneider, M.B.; Ulmschneider, J.P. Spontaneous Formation of Structurally Diverse Membrane Channel Architectures from a Single Antimicrobial Peptide. Nat. Commun. 2016, 7, 13535. [Google Scholar] [CrossRef]
- Santo, K.P.; Irudayam, S.J.; Berkowitz, M.L. Melittin Creates Transient Pores in a Lipid Bilayer: Results from Computer Simulations. J. Phys. Chem. B 2013, 117, 5031–5042. [Google Scholar] [CrossRef]
- Lu, N.; Yang, K.; Yuan, B.; Ma, Y. Molecular Response and Cooperative Behavior during the Interactions of Melittin with a Membrane: Dissipative Quartz Crystal Microbalance Experiments and Simulations. J. Phys. Chem. B 2012, 116, 9432–9438. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Im, W. Automated Builder and Database of Protein/Membrane Complexes for Molecular Dynamics Simulations. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Zhou, H.X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- De Jesus, A.J.; Allen, T.W. The Role of Tryptophan Side Chains in Membrane Protein Anchoring and Hydrophobic Mismatch. Biochim. Biophys. Acta Biomembr. 2013, 1828, 864–876. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Chodera, J.D. Statistically Optimal Analysis of Samples from Multiple Equilibrium States. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef]
- McGibbon, R.; Beauchamp, K.; Harrigan, M.; Klein, C.; Swails, J.; Hernández, C.; Schwantes, C.; Wang, L.P.; Lane, T.; Pande, V. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.; Sheng, Y.; Wang, J.; Wang, W. Chirality-Dependent Adsorption between Amphipathic Peptide and POPC Membrane. Int. J. Mol. Sci. 2019, 20, 4760. https://doi.org/10.3390/ijms20194760
Chen K, Sheng Y, Wang J, Wang W. Chirality-Dependent Adsorption between Amphipathic Peptide and POPC Membrane. International Journal of Molecular Sciences. 2019; 20(19):4760. https://doi.org/10.3390/ijms20194760
Chicago/Turabian StyleChen, Ke, Yuebiao Sheng, Jun Wang, and Wei Wang. 2019. "Chirality-Dependent Adsorption between Amphipathic Peptide and POPC Membrane" International Journal of Molecular Sciences 20, no. 19: 4760. https://doi.org/10.3390/ijms20194760
APA StyleChen, K., Sheng, Y., Wang, J., & Wang, W. (2019). Chirality-Dependent Adsorption between Amphipathic Peptide and POPC Membrane. International Journal of Molecular Sciences, 20(19), 4760. https://doi.org/10.3390/ijms20194760