Proteasome Inhibitors Suppress ErbB Family Expression through HSP90-Mediated Lysosomal Degradation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
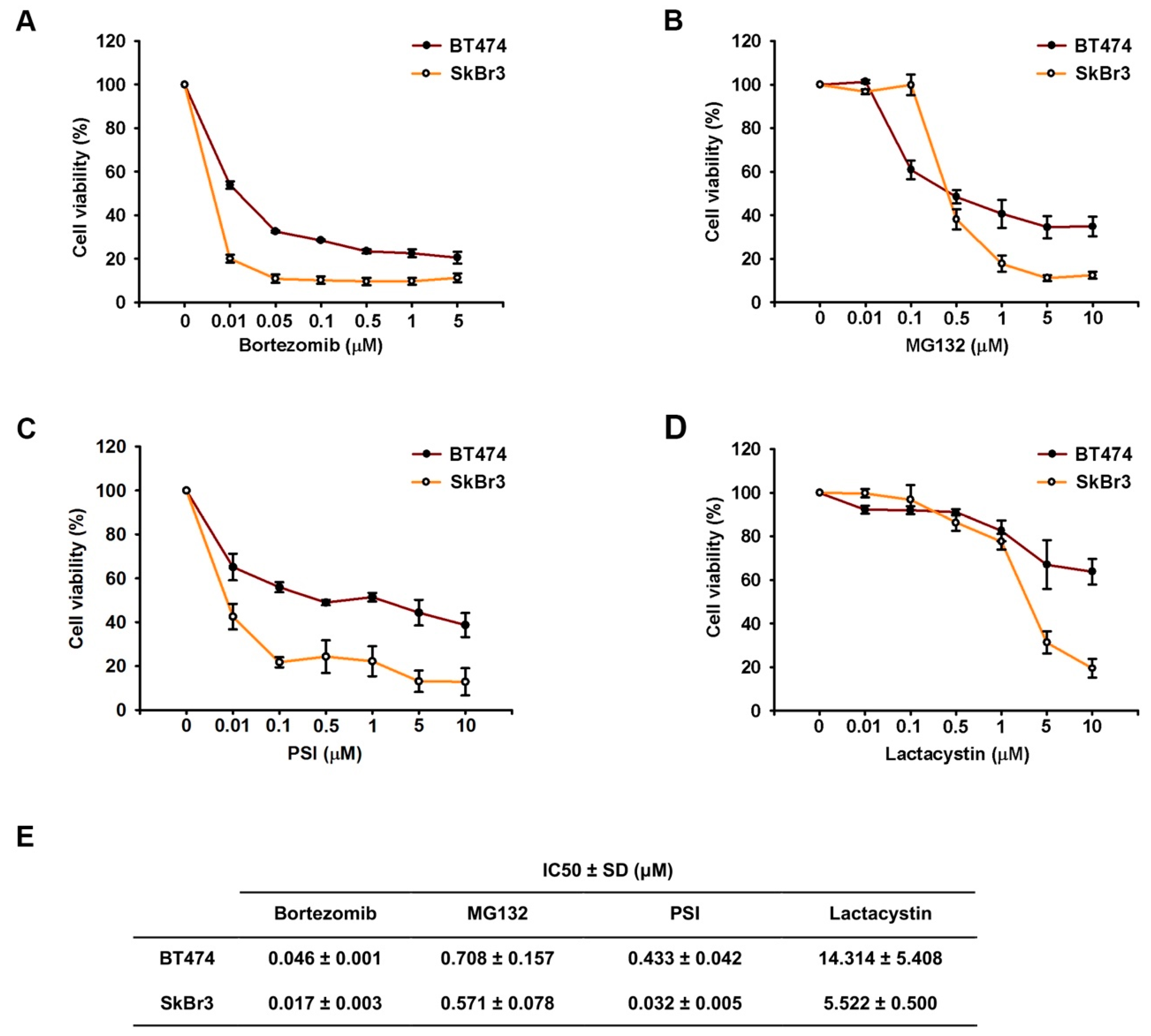
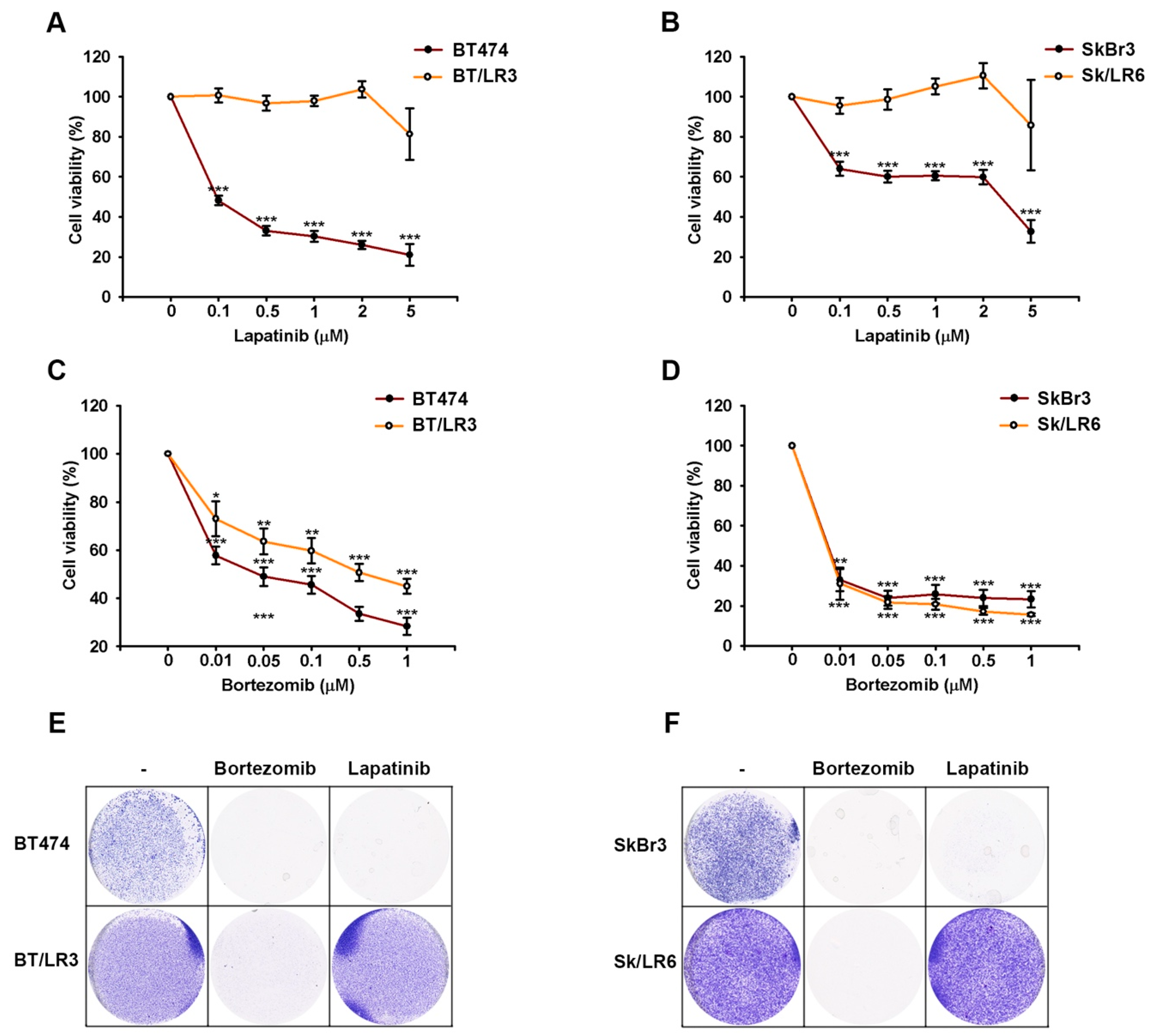
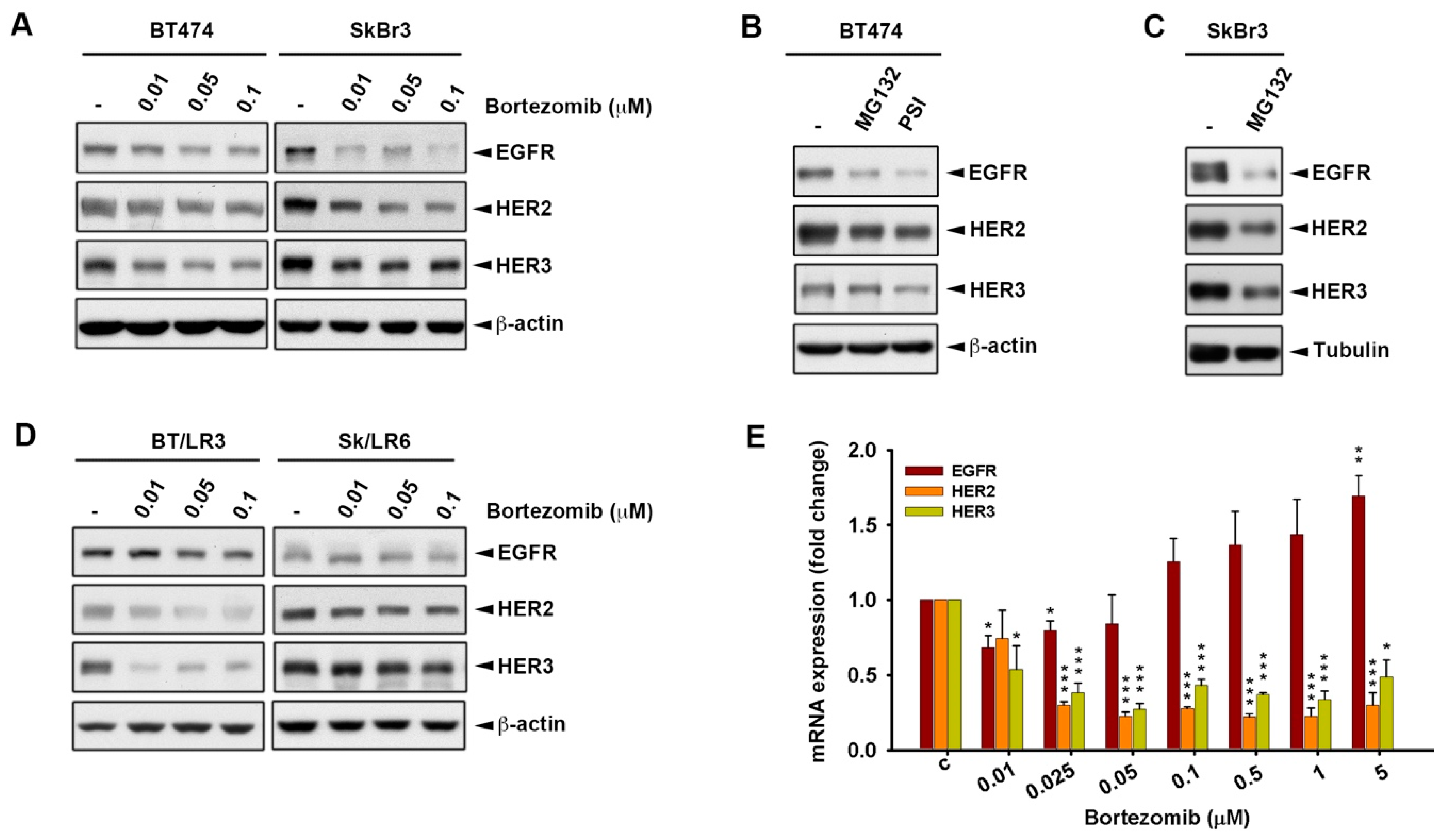
2.1. Proteasome Inhibitors Reduced Cell Growth by Suppressing Expressions of ErbB Family Schemes
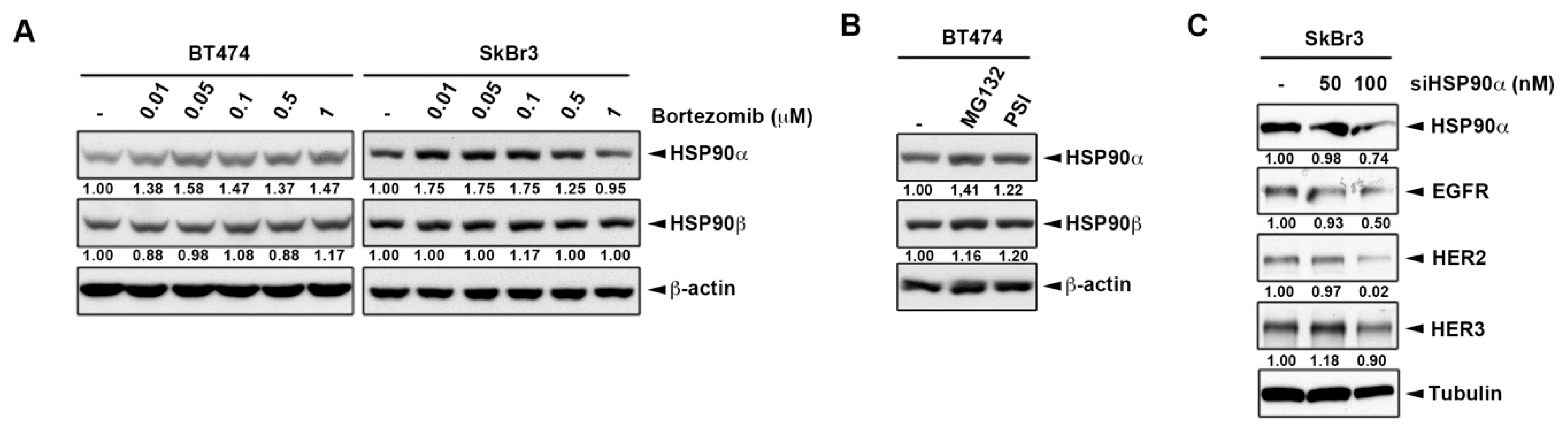
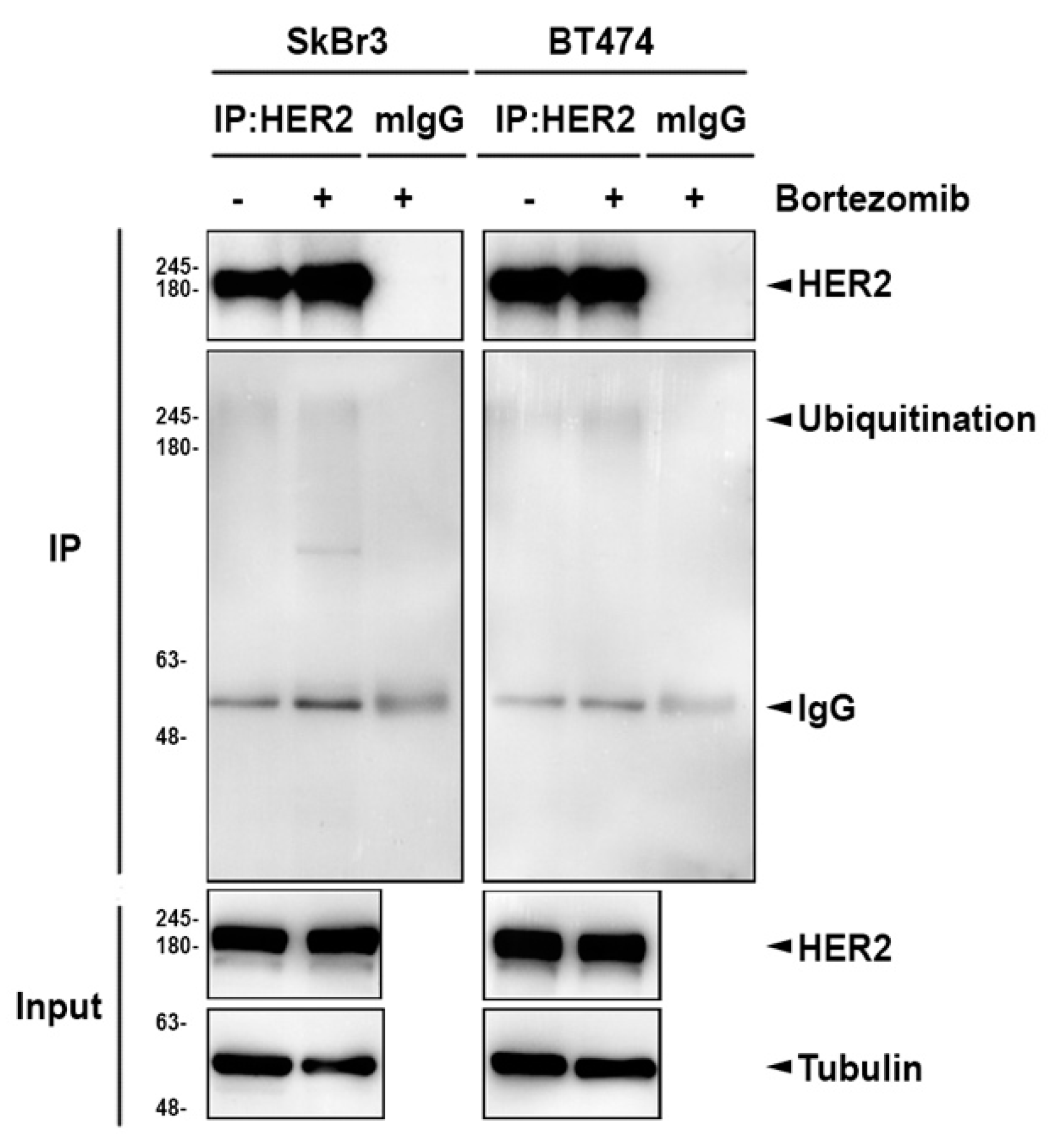
2.2. Inhibition of Heat Shock Protein HSP90 Mediates the Proteasome Inhibitor-Induced ErbB Family Degradation
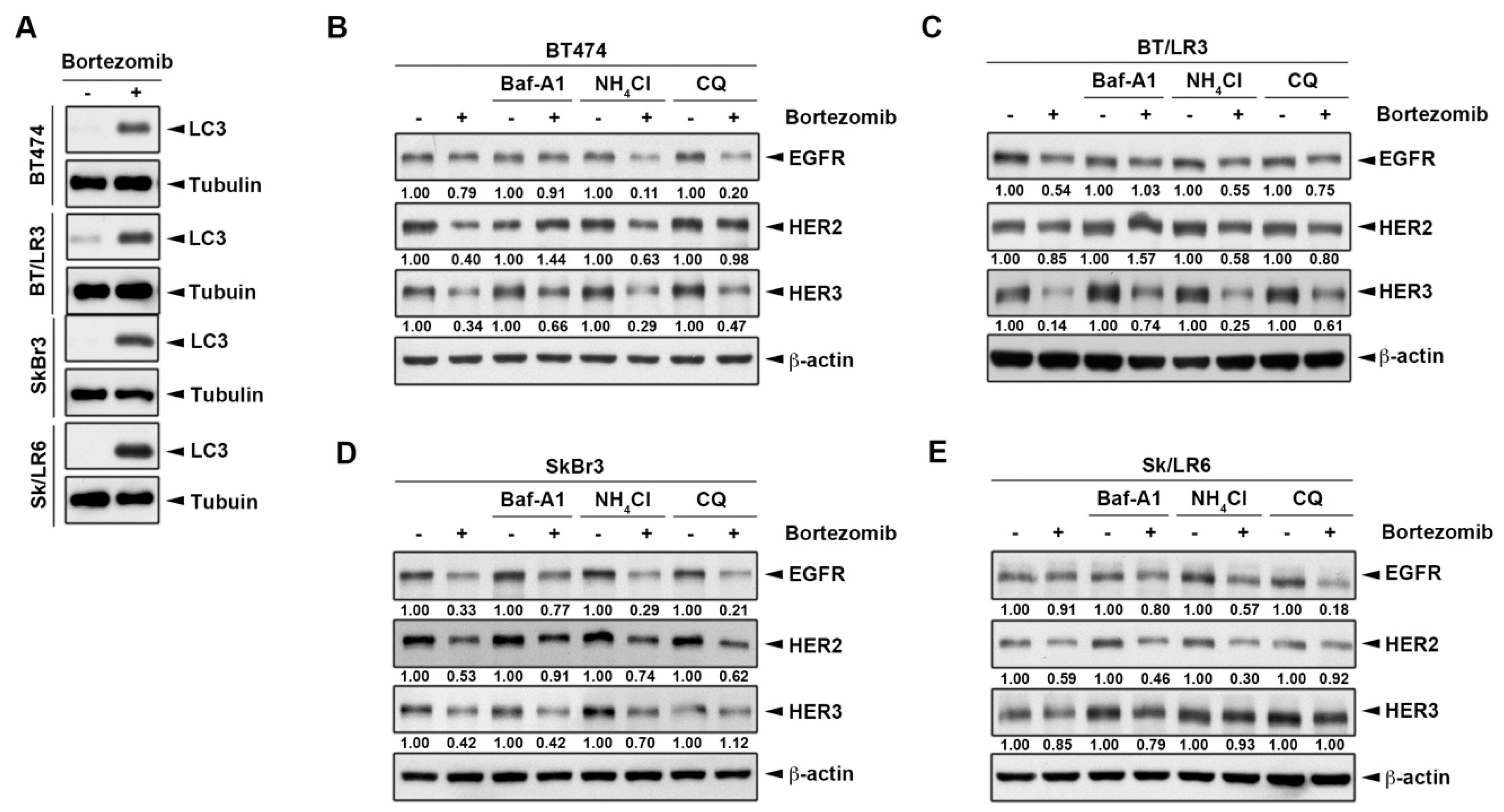
2.3. The Lysosomal Pathway is Involved in Bortezomib-Induced ErbB Degradation
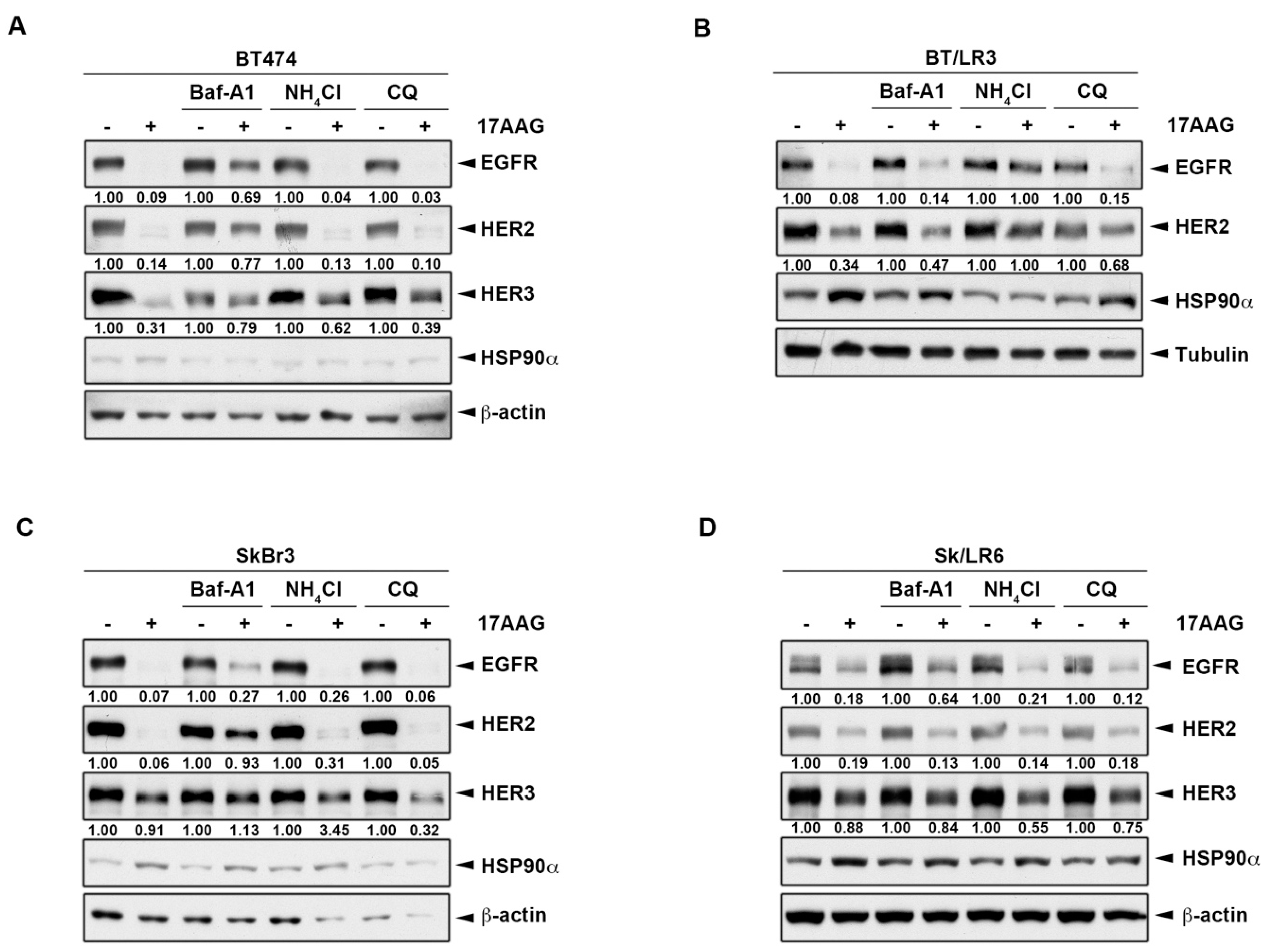
2.4. HSP90 Inhibitors Also Downregulated ErbB Family Expressions via the Lysosomal Pathway
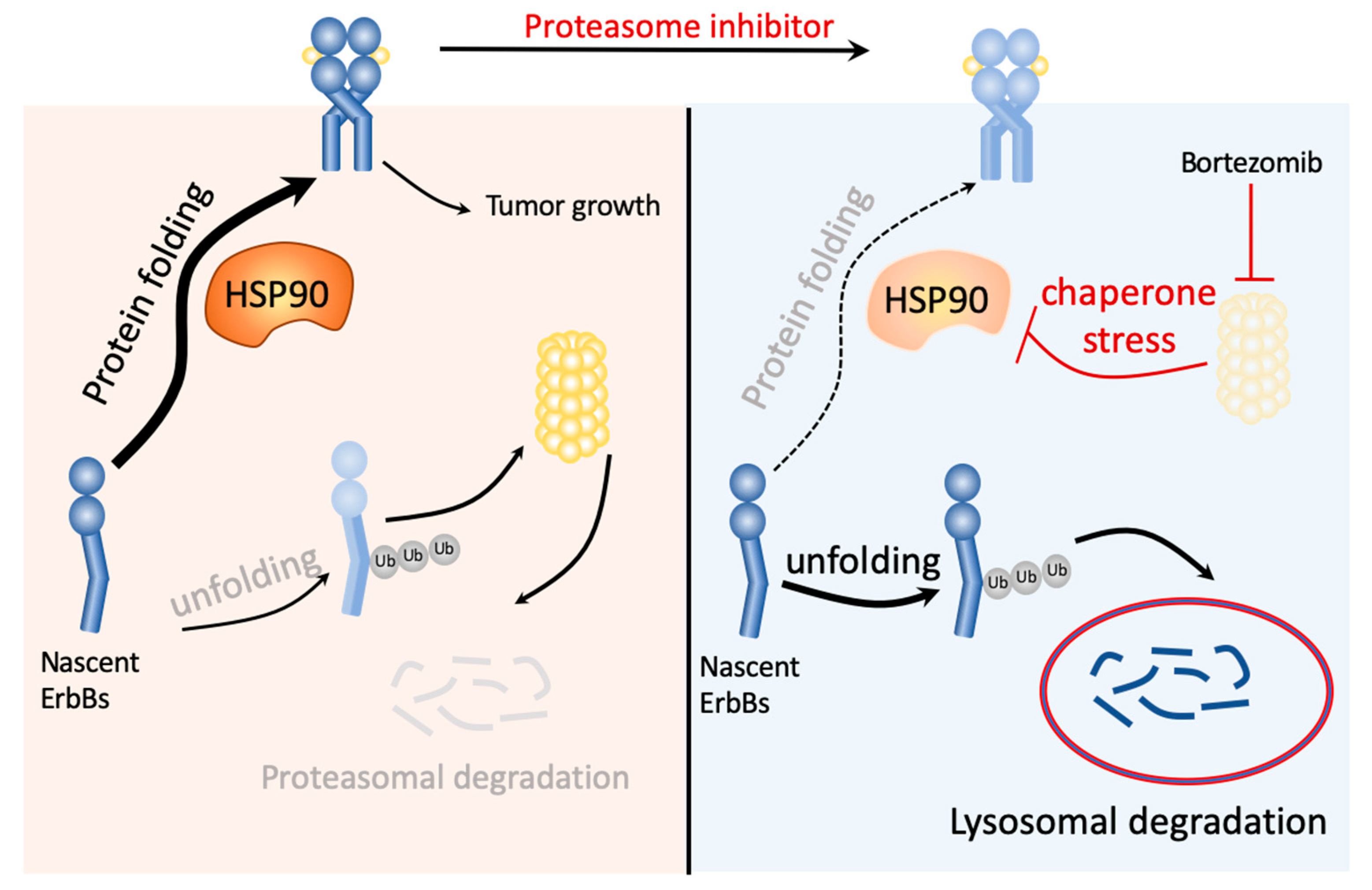
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Protein Extraction and Western Blot Analysis
4.3. MTT Assay
4.4. Clonogenic Assay
4.5. RNA Extraction and Quantitative Reverse Transcription PCR
4.6. RNA Interference
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal Growth Factor Receptor |
| PI3K/Akt | Phosphatidylinositol 3-kinase/Protein Kinase B |
| MAPK/Erk | Mitogen-Activated Protein Kinases/Extracellular signal-Related kinases |
| NF-κB | Nuclear Factor kappa light chain enhancer of activated B cells |
| HSP90 | Heat Shock Protein 90 |
| Baf-A1 | Bafilomycin A1 |
| NH4Cl | Ammonium chloride |
| CQ | Chloroquine diphosphate |
References
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar]
- Grandis, J.R.; Sok, J.C. Signaling through the epidermal growth factor receptor during the development of malignancy. Pharmacol. Ther. 2004, 102, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the her-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, V.; Revillion, F.; Hebbar, M.; Hornez, L.; Peyrat, J.P. Prognostic value of the type i growth factor receptors in a large series of human primary breast cancers quantified with a real-time reverse transcription-polymerase chain reaction assay. Clin. Cancer Res. 2000, 6, 4217–4225. [Google Scholar] [PubMed]
- Way, T.D.; Lin, J.K. Role of her2/her3 co-receptor in breast carcinogenesis. Future Oncol. 2005, 1, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Wallasch, C.; Weiss, F.U.; Niederfellner, G.; Jallal, B.; Issing, W.; Ullrich, A. Heregulin-dependent regulation of her2/neu oncogenic signaling by heterodimerization with her3. EMBO J. 1995, 14, 4267–4275. [Google Scholar] [CrossRef]
- Tovey, S.M.; Witton, C.J.; Bartlett, J.M.; Stanton, P.D.; Reeves, J.R.; Cooke, T.G. Outcome and human epidermal growth factor receptor (her) 1-4 status in invasive breast carcinomas with proliferation indices evaluated by bromodeoxyuridine labelling. Breast Cancer Res. 2004, 6, R246–R251. [Google Scholar] [CrossRef] [PubMed]
- Nuciforo, P.; Radosevic-Robin, N.; Ng, T.; Scaltriti, M. Quantification of her family receptors in breast cancer. Breast Cancer Res. 2015, 17, 53. [Google Scholar] [CrossRef]
- Padma, V.V. An overview of targeted cancer therapy. Biomedicine 2015, 5, 19. [Google Scholar] [CrossRef]
- Spector, N.L.; Xia, W.; Burris, H., 3rd; Hurwitz, H.; Dees, E.C.; Dowlati, A.; O’Neil, B.; Overmoyer, B.; Marcom, P.K.; Blackwell, K.L.; et al. Study of the biologic effects of lapatinib, a reversible inhibitor of erbb1 and erbb2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 2502–2512. [Google Scholar] [CrossRef]
- Chen, F.L.; Xia, W.; Spector, N.L. Acquired resistance to small molecule erbb2 tyrosine kinase inhibitors. Clin. Cancer Res. 2008, 14, 6730–6734. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chen, K.B.; Tsai, C.H.; Tsai, F.J.; Huang, C.Y.; Tang, C.H.; Yang, J.S.; Hsu, Y.M.; Peng, S.F.; Chung, J.G. Casticin inhibits human prostate cancer du 145 cell migration and invasion via ras/akt/nf-kappab signaling pathways. J. Food Biochem. 2019, 43, e12902. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. Nf-kappab at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. Nf-kappa b activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [PubMed]
- Barkett, M.; Gilmore, T.D. Control of apoptosis by rel/nf-kappab transcription factors. Oncogene 1999, 18, 6910–6924. [Google Scholar] [CrossRef] [PubMed]
- Kanarek, N.; London, N.; Schueler-Furman, O.; Ben-Neriah, Y. Ubiquitination and degradation of the inhibitors of nf-kappab. Cold Spring Harb. Perspect Biol. 2010, 2, a000166. [Google Scholar] [CrossRef]
- Frankland-Searby, S.; Bhaumik, S.R. The 26s proteasome complex: An attractive target for cancer therapy. Biochim. Biophys. Acta 2012, 1825, 64–76. [Google Scholar] [CrossRef]
- Chen, Y.J.; Yeh, M.H.; Yu, M.C.; Wei, Y.L.; Chen, W.S.; Chen, J.Y.; Shih, C.Y.; Tu, C.Y.; Chen, C.H.; Hsia, T.C.; et al. Lapatinib-induced nf-kappab activation sensitizes triple-negative breast cancer cells to proteasome inhibitors. Breast Cancer Res. 2013, 15, R108. [Google Scholar] [CrossRef]
- Ciechanover, A.; Schwartz, A.L. The ubiquitin-proteasome pathway: The complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. USA 1998, 95, 2727–2730. [Google Scholar] [CrossRef]
- Sanchez-Serrano, I. Success in translational research: Lessons from the development of bortezomib. Nat. Rev. Drug Discov. 2006, 5, 107–114. [Google Scholar] [CrossRef]
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (velcadetrade mark) in the treatment of multiple myeloma. Ther. Clin Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef]
- Zhang, L.; Fok, J.H.; Davies, F.E. Heat shock proteins in multiple myeloma. Oncotarget 2014, 5, 1132–1148. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Nooka, A.K.; Jaye, D.L.; Bahlis, N.J.; Lonial, S.; Boise, L.H. Bortezomib-induced heat shock response protects multiple myeloma cells and is activated by heat shock factor 1 serine 326 phosphorylation. Oncotarget 2016, 7, 59727–59741. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C. Mechanisms of hsp90 regulation. Biochem. J. 2016, 473, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Sawai, A.; Chandarlapaty, S.; Greulich, H.; Gonen, M.; Ye, Q.; Arteaga, C.L.; Sellers, W.; Rosen, N.; Solit, D.B. Inhibition of hsp90 down-regulates mutant epidermal growth factor receptor (egfr) expression and sensitizes egfr mutant tumors to paclitaxel. Cancer Res. 2008, 68, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Mayor-Lopez, L.; Tristante, E.; Carballo-Santana, M.; Carrasco-Garcia, E.; Grasso, S.; Garcia-Morales, P.; Saceda, M.; Lujan, J.; Garcia-Solano, J.; Carballo, F.; et al. Comparative study of 17-aag and nvp-auy922 in pancreatic and colorectal cancer cells: Are there common determinants of sensitivity? Transl. Oncol. 2014, 7, 590–604. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.D.; Da Pieve, C.; Burley, T.A.; Smith, R.; Ciobota, D.M.; Allott, L.; Harrington, K.J.; Oyen, W.J.G.; Smith, G.; Kramer-Marek, G. Her3-mediated resistance to hsp90 inhibition detected in breast cancer xenografts by affibody-based pet imaging. Clin. Cancer Res. 2018, 24, 1853–1865. [Google Scholar] [CrossRef]
- Ma, C.; Niu, X.; Luo, J.; Shao, Z.; Shen, K. Combined effects of lapatinib and bortezomib in human epidermal receptor 2 (her2)-overexpressing breast cancer cells and activity of bortezomib against lapatinib-resistant breast cancer cells. Cancer Sci. 2010, 101, 2220–2226. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Tapia, M.A.; Bosch, M.; Oliva, C.; Domingo-Domenech, J.; Mellado, B.; Rolfe, M.; Ross, J.S.; Gascon, P.; Rovira, A.; et al. Differential cellular and molecular effects of bortezomib, a proteasome inhibitor, in human breast cancer cells. Mol. Cancer Ther. 2006, 5, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Sridhara, R.; Pazdur, R. United states food and drug administration approval summary: Bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin. Cancer Res. 2006, 12, 2955–2960. [Google Scholar] [CrossRef]
- Kane, R.C.; Bross, P.F.; Farrell, A.T.; Pazdur, R. Velcade: U.S. Fda approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist 2003, 8, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Melman, L.; Geuze, H.J.; Li, Y.; McCormick, L.M.; Van Kerkhof, P.; Strous, G.J.; Schwartz, A.L.; Bu, G. Proteasome regulates the delivery of ldl receptor-related protein into the degradation pathway. Mol. Biol. Cell 2002, 13, 3325–3335. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berges, C.; Haberstock, H.; Fuchs, D.; Miltz, M.; Sadeghi, M.; Opelz, G.; Daniel, V.; Naujokat, C. Proteasome inhibition suppresses essential immune functions of human cd4+ t cells. Immunology 2008, 124, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Klikova, K.; Stefanikova, A.; Pilchova, I.; Hatok, J.; Chudy, P.; Chudej, J.; Dobrota, D.; Racay, P. Differential impact of bortezomib on hl-60 and k562 cells. Gen. Physiol. Biophys. 2015, 34, 33–42. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Citri, A.; Gan, J.; Mosesson, Y.; Vereb, G.; Szollosi, J.; Yarden, Y. Hsp90 restrains erbb-2/her2 signalling by limiting heterodimer formation. EMBO Rep. 2004, 5, 1165–1170. [Google Scholar] [CrossRef]
- Berezowska, S.; Novotny, A.; Bauer, K.; Feuchtinger, A.; Slotta-Huspenina, J.; Becker, K.; Langer, R.; Walch, A. Association between hsp90 and her2 in gastric and gastroesophageal carcinomas. PLoS ONE 2013, 8, e69098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Burrows, F. Targeting multiple signal transduction pathways through inhibition of hsp90. J. Mol. Med. 2004, 82, 488–499. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, P.; Lovchik, M.A.; Li, Y.; Tang, L.; Chen, Z.; Zeng, R.; Ma, D.; Yuan, J.; Yu, Q. Cyclodepsipeptide toxin promotes the degradation of hsp90 client proteins through chaperone-mediated autophagy. J. Cell Biol. 2009, 185, 629–639. [Google Scholar] [CrossRef]
- Weber, H.; Valbuena, J.R.; Barbhuiya, M.A.; Stein, S.; Kunkel, H.; Garcia, P.; Bizama, C.; Riquelme, I.; Espinoza, J.A.; Kurtz, S.E.; et al. Small molecule inhibitor screening identifified hsp90 inhibitor 17-aag as potential therapeutic agent for gallbladder cancer. Oncotarget 2017, 8, 26169–26184. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Kalmar, E.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Ohta, T.; Fukuda, M. Ubiquitin and breast cancer. Oncogene 2004, 23, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Lipkowitz, S. The role of the ubiquitination-proteasome pathway in breast cancer: Ubiquitin mediated degradation of growth factor receptors in the pathogenesis and treatment of cancer. Breast Cancer Res. 2003, 5, 8–15. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.D.; Young, M.R. Ammonium chloride, an inhibitor of phagosome-lysosome fusion in macrophages, concurrently induces phagosome-endosome fusion, and opens a novel pathway: Studies of a pathogenic mycobacterium and a nonpathogenic yeast. J. Exp. Med. 1991, 174, 881–889. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toft, D.O.; Ames, M.M.; Erlichman, C. The hsp90 chaperone complex as a novel target for cancer therapy. Ann. Oncol. 2003, 14, 1169–1176. [Google Scholar] [CrossRef]
- Solit, D.B.; Zheng, F.F.; Drobnjak, M.; Munster, P.N.; Higgins, B.; Verbel, D.; Heller, G.; Tong, W.; Cordon-Cardo, C.; Agus, D.B.; et al. 17-allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and her-2/neu and inhibits the growth of prostate cancer xenografts. Clin. Cancer Res. 2002, 8, 986–993. [Google Scholar]
- Wang, D.; Xu, Q.; Yuan, Q.; Jia, M.; Niu, H.; Liu, X.; Zhang, J.; Young, C.Y.; Yuan, H. Proteasome inhibition boosts autophagic degradation of ubiquitinated-agr2 and enhances the antitumor efficiency of bevacizumab. Oncogene 2019, 38, 3458–3474. [Google Scholar] [CrossRef]
- Wang, A.; Bao, Y.; Wu, Z.; Zhao, T.; Wang, D.; Shi, J.; Liu, B.; Sun, S.; Yang, F.; Wang, L.; et al. Long noncoding rna egfr-as1 promotes cell growth and metastasis via affecting hur mediated mrna stability of egfr in renal cancer. Cell Death Dis. 2019, 10, 154. [Google Scholar] [CrossRef]
- Kandasamy, K.; Kraft, A.S. Proteasome inhibitor ps-341 (velcade) induces stabilization of the trail receptor dr5 mrna through the 3’-untranslated region. Mol. Cancer Ther. 2008, 7, 1091–1100. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Mimnaugh, E.G.; Xu, W.; Vos, M.; Yuan, X.; Isaacs, J.S.; Bisht, K.S.; Gius, D.; Neckers, L. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol. Cancer Ther. 2004, 3, 551–566. [Google Scholar]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010, 584, 1393–1398. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Chowdhury, R.D. Molecular crosstalk between the proteasome, aggresomes and autophagy: Translational potential and clinical implications. Cancer Lett. 2012, 325, 147–154. [Google Scholar] [CrossRef]
- Aligayer, H.; Boyd, D.D.; Heiss, M.M.; Abdalla, E.K.; Curley, S.A.; Gallick, G.E. Activation of src kinase in primary colorectal carcinoma: An indicator of poor clinical prognosis. Cancer 2002, 94, 344–351. [Google Scholar] [CrossRef]
- Erpel, T.; Courtneidge, S.A. Src family protein tyrosine kinases and cellular signal transduction pathways. Curr. Opin Cell Biol. 1995, 7, 176–182. [Google Scholar] [CrossRef]
- Summy, J.M.; Gallick, G.E. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003, 22, 337–358. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular functions regulated by src family kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef]
- Bao, J.; Gur, G.; Yarden, Y. Src promotes destruction of c-cbl: Implications for oncogenic synergy between src and growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 2438–2443. [Google Scholar] [CrossRef]
- Dong, Y.; Liang, C.; Zhang, B.; Ma, J.; He, X.; Chen, S.; Zhang, X.; Chen, W. Bortezomib enhances the therapeutic efficacy of dasatinib by promoting c-KIT internalization-induced apoptosis in gastrointestinal stromal tumor cells. Cancer Lett. 2015, 361, 137–146. [Google Scholar] [CrossRef]
- Mocciaro, A.; Rape, M. Emerging regulatory mechanisms in ubiquitin-dependent cell cycle control. J. Cell Sci. 2012, 125, 255–263. [Google Scholar] [CrossRef]
- Huang, W.C.; Hung, C.M.; Wei, C.T.; Chen, T.M.; Chien, P.H.; Pan, H.L.; Lin, Y.M.; Chen, Y.J. Interleukin-6 expression contributes to lapatinib resistance through maintenance of stemness property in her2-positive breast cancer cells. Oncotarget 2016, 7, 62352–62363. [Google Scholar] [CrossRef]
- Wu, K.M.; Hsu, Y.M.; Ying, M.C.; Tsai, F.J.; Tsai, C.H.; Chung, J.G.; Yang, J.S.; Tang, C.H.; Cheng, L.Y.; Su, P.H.; et al. High-density lipoprotein ameliorates palmitic acid-induced lipotoxicity and oxidative dysfunction in h9c2 cardiomyoblast cells via ros suppression. Nutr. Metab. 2019, 16, 36. [Google Scholar] [CrossRef]
- Lee, H.P.; Chen, P.C.; Wang, S.W.; Fong, Y.C.; Tsai, C.H.; Tsai, F.J.; Chung, J.G.; Huang, C.Y.; Yang, J.S.; Hsu, Y.M.; et al. Plumbagin suppresses endothelial progenitor cell-related angiogenesis in vitro and in vivo. J. Funct. Foods 2019, 52, 537–544. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huynh, T.K.; Ho, C.-Y.; Tsai, C.-H.; Wang, C.-K.; Chen, Y.-J.; Bau, D.-T.; Tu, C.-Y.; Li, T.-S.; Huang, W.-C. Proteasome Inhibitors Suppress ErbB Family Expression through HSP90-Mediated Lysosomal Degradation. Int. J. Mol. Sci. 2019, 20, 4812. https://doi.org/10.3390/ijms20194812
Huynh TK, Ho C-Y, Tsai C-H, Wang C-K, Chen Y-J, Bau D-T, Tu C-Y, Li T-S, Huang W-C. Proteasome Inhibitors Suppress ErbB Family Expression through HSP90-Mediated Lysosomal Degradation. International Journal of Molecular Sciences. 2019; 20(19):4812. https://doi.org/10.3390/ijms20194812
Chicago/Turabian StyleHuynh, Thanh Kieu, Chien-Yi Ho, Chi-Hua Tsai, Chien-Kuo Wang, Yun-Ju Chen, Da-Tian Bau, Chih-Yen Tu, Tzong-Shiun Li, and Wei-Chien Huang. 2019. "Proteasome Inhibitors Suppress ErbB Family Expression through HSP90-Mediated Lysosomal Degradation" International Journal of Molecular Sciences 20, no. 19: 4812. https://doi.org/10.3390/ijms20194812
APA StyleHuynh, T. K., Ho, C.-Y., Tsai, C.-H., Wang, C.-K., Chen, Y.-J., Bau, D.-T., Tu, C.-Y., Li, T.-S., & Huang, W.-C. (2019). Proteasome Inhibitors Suppress ErbB Family Expression through HSP90-Mediated Lysosomal Degradation. International Journal of Molecular Sciences, 20(19), 4812. https://doi.org/10.3390/ijms20194812