Metabolomic Perspectives in Antiblastic Cardiotoxicity and Cardioprotection

 ,
,  ,
, 

Abstract
1. Introduction
2. Metabolic Derangements in Heart Failure (HF)
3. Experimental Techniques Used in Metabolomics Studies
4. Metabolomic Studies in Cardiotoxicity and Cardioprotection
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| CTX | Cardiotoxicity |
| ATP | Adenosine TriPhosphate |
| PDH | Pyruvate DeHydrogenase |
| DCA | DiChloreAcetate |
| CoA | CoEnzyme A |
| CPTI | Carnitine Palmitoyl Transferase I |
| FA | Fatty Acids |
| HF | Heart Failure |
| LC 3-KAT | Long-Chain 3-KetoAcyl-coA Thiolsase |
| ROS | Reactive Oxygen Species |
| MCD | Malonyl-CoA Decarboxylase |
| ADP | Adenosine DiPhosphate |
| PPAR-γ | Proliferator-Activated Receptor-γ |
| PGC-1α | Proliferator-activated receptor-γ Coactivator 1α |
| BNP | Brain Natriuretic Peptide |
| UCPs | Mitochondrial Uncoupling Proteins |
| AMP | Adenosine MonoPhosphate |
| CK | Creatine Kinase |
| AICAR | 5′-AminoImidazole-4-CarboxyAmide-Ribonucleoside |
| AMPK | Adenosine-MonoPhosphate Kinase |
| GLUTs | Glucose Transporters |
| NMR | Nuclear Magnetic Resonance |
| MS | Mass Spectrometry |
| GC | Gas Chromatography |
| LC | Liquid Chromatograph |
| HR-MAS-NMR | High-resolution magic-angle-spinning NMR |
| DOX | Doxorubicine |
| iNOS | inducible Nitric Oxide Synthases |
| eNOS | endothelial Nitric Oxide Synthases |
| THP | pirarubicin |
| L-THP | Liposome pirarubicin |
| F-THP | Free pirarubicin |
| CY | Cyclophosphamide |
| UPLC–Q-TOF-MS | Ultra-Performance Liquid Chromatography–Quadrupole Time-of-Flight MS |
| 1H-NMR | Hydrogen NMR |
| hiPSC-CMs | Human induced Pluripotent Stem Cell-Derived CardioMyocytes |
| DZR | Dexrazoxane |
| TKIs | Tyrosine Kinase Inhibitors |
| HR-MAS NMR | High-Resolution Magic-Angle-Spinning NMR |
References
- Mele, D.; Nardozza, M.; Spallarossa, P.; Frassoldati, A.; Tocchetti, C.G.; Cadeddu, C.; Madonna, R.; Malagù, M.; Ferrari, R.; Mercuro, G. Current views on anthracycline cardiotoxicity. Heart Fail. Rev. 2016, 21, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Cadeddu, C.; Piras, A.; Dessì, M.; Madeddu, C.; Mantovani, G.; Scartozzi, M.; Hagendorff, A.; Colonna, P.; Mercuro, G. Timing of the negative effects of trastuzumab on cardiac mechanics after anthracycline chemotherapy. Int. J. Cardiovasc. Imaging 2017, 33, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Madeddu, C.; Deidda, M.; Piras, A.; Cadeddu, C.; Demurtas, L.; Puzzoni, M.; Piscopo, G.; Scartozzi, M.; Mercuro, G. Pathophysiology of cardiotoxicity induced by nonanthracycline chemotherapy. J. Cardiovasc. Med. (Hagerstown) 2016, 17 (Suppl. S1), S12–S18. [Google Scholar] [CrossRef] [PubMed]
- Mercuro, G.; Cadeddu, C.; Piras, A.; Dessì, M.; Madeddu, C.; Deidda, M.; Serpe, R.; Massa, E.; Mantovani, G. Early epirubicin-induced myocardial dysfunction revealed by serial tissue doppler echocardiography: Correlation with inflammatory and oxidative stress markers. Oncologist 2007, 12, 1124–1133. [Google Scholar] [CrossRef]
- Deidda, M.; Madonna, R.; Mango, R.; Pagliaro, P.; Bassareo, P.P.; Cugusi, L.; Romano, S.; Penco, M.; Romeo, F.; Mercuro, G. Novel insights in pathophysiology of antiblastic drugs-induced cardiotoxicity and cardioprotection. J. Cardiovasc. Med. (Hagerstown) Spec. Issue Cardiotoxic. Antiblast. Drugs Cardioprot. 2016, 17 (Suppl. S1), e76–e83. [Google Scholar] [CrossRef] [PubMed]
- Deidda, M.; Piras, C.; Bassareo, P.P.; Cadeddu Dessalvi, C.; Mercuro, G. Metabolomics, a promising approach to translational research in cardiology. IJC Metab. Endocr. 2015, 9, 31–38. [Google Scholar] [CrossRef]
- Deidda, M.; Piras, C.; Dessalvi, C.C.; Locci, E.; Barberini, L.; Torri, F.; Ascedu, F.; Atzori, L.; Mercuro, G. Metabolomic approach to profile functional and metabolic changes in heart failure. J. Transl. Med. 2015, 13, 297. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2017, 356, 1140–1151. [Google Scholar] [CrossRef]
- Osorio, J.C.; Stanley, W.C.; Linke, A.; Castellari, M.; Diep, Q.N.; Panchal, A.R.; Hintze, T.H.; Lopaschuk, G.D.; Recchia, F.A. Impaired myocardial fatty acid oxidation and reduced protein expression of retinoid X receptor-alpha in pacing-induced heart failure. Circulation 2002, 106, 606–612. [Google Scholar] [CrossRef]
- Chandler, M.P.; Kerner, J.; Huang, H.; Vazquez, E.; Reszko, A.; Martini, W.Z.; Hoppel, C.L.; Imai, M.; Rastogi, S.; Sabbah, H.N.; et al. Moderate severity heart failure does not involve a downregulation of myocardial fatty acid oxidation. Am. J. Physiol Heart Circ. Physiol. 2004, 287, H1538–H1543. [Google Scholar] [CrossRef]
- Nascimben, L.; Ingwall, J.S.; Lorell, B.H.; Pinz, I.; Schultz, V.; Tornheim, K.; Tian, R. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 2004, 44, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Wallhaus, T.R.; Degrado, T.R.; Russell, D.C.; Stanko, P.; Nickles, R.J.; Stone, C.K. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F]fluoro-6-thia-heptadecanoic acid and [18F]FDG in patients with congestive heart failure. J. Nucl. Med. 2001, 42, 55–62. [Google Scholar] [PubMed]
- Fillmore, N.; Lopaschuk, G.D. Targeting mitochondrial oxidative metabolism as an approach to treat heart failure. Biochim. Biophys. Acta 2013, 1833, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Sallustio, B.; Horowitz, J.D. Drugs that affect cardiac metabolism: Focus on perhexiline. Cardiovasc. Drugs Ther. 2016, 30, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Revenco, D.; Morgan, J.P. Metabolic modulation and cellular therapy of cardiac dysfunction and failure. J. Cell. Mol. Med. 2009, 13, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Noordali, H.; Loudon, B.L.; Frenneaux, M.P.; Madhani, M. Cardiac metabolism—A promising therapeutic target for heart failure. Pharmacol. Ther. 2018, 182, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Kantor, P.F.; Lucien, A.; Kozak, R.; Lopaschuk, G.D. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ. Res. 2000, 86, 580–588. [Google Scholar] [CrossRef]
- Lee, L.; Horowitz, J.; Frenneaux, M. Metabolic manipulation in ischaemic heart disease, a novel approach to treatment. Eur. Heart J. 2004, 25, 634–641. [Google Scholar] [CrossRef]
- Li, Y.J.; Wang, P.H.; Chen, C.; Zou, M.H.; Wang, D.W. Improvement of mechanical heart function by trimetazidine in db/db mice. Acta Pharmacol. Sin. 2010, 31, 560–569. [Google Scholar] [CrossRef]
- Cheng, J.F.; Huang, Y.; Penuliar, R.; Nishimoto, M.; Liu, L.; Arrhenius, T.; Yang, G.; O’leary, E.; Barbosa, M.; Barr, R.; et al. Discovery of potent and orally available malonyl-coa decarboxylase inhibitors as cardioprotective agents. J. Med. Chem. 2006, 49, 4055–4058. [Google Scholar] [CrossRef]
- Wu, H.; Zhu, Q.; Cai, M.; Tong, X.; Liu, D.; Huang, J.; Yang, G.; Jiang, Y. Effect of inhibiting malonyl-coa decarboxylase on cardiac remodelling after myocardial infarction in rats. Cardiology 2014, 127, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Quigley, A.F.; Kapsa, R.M.; Esmore, D.; Hale, G.; Byrne, E. Mitochondrial respiratory chain activity in idiopathic dilated cardiomyopathy. J. Card. Fail. 2000, 6, 47–55. [Google Scholar] [CrossRef]
- Vega, R.B.; Huss, J.M.; Kelly, D.P. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 2000, 20, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Marin-Garcia, J.; Goldenthal, M.J.; Moe, G.W. Mitochondrial pathology in cardiac failure. Cardiovasc. Res. 2001, 49, 17–26. [Google Scholar] [CrossRef]
- Lehman, J.J.; Kelly, D.P. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin. Exp. Pharmacol. Physiol. 2002, 29, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy metabolism in heart failure. J. Physiol. 2004, 555, 1–13. [Google Scholar] [CrossRef]
- Arany, Z.; Novikov, M.; Chin, S.; Ma, Y.; Rosenzweig, A.; Spiegelman, B.M. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 10086–10091. [Google Scholar] [CrossRef]
- Li, P.; Luo, S.; Pan, C.; Cheng, X. Modulation of fatty acid metabolism is involved in the alleviation of isoproterenol-induced rat heart failure by fenofibrate. Mol. Med. Rep. 2015, 12, 7899–7906. [Google Scholar] [CrossRef][Green Version]
- Labinskyy, V.; Bellomo, M.; Chandler, M.P.; Young, M.E.; Lionetti, V.; Qanud, K.; Bigazzi, F.; Sampietro, T.; Stanley, W.C.; Recchia, F.A. Chronic activation of peroxisome proliferator-activated receptor-alpha with fenofibrate prevents alterations in cardiac metabolic phenotype without changing the onset of decompensation in pacing-induced heart failure. J. Pharmacol. Exp. Ther. 2007, 321, 165–171. [Google Scholar] [CrossRef]
- Akhmedov, A.T.; Rybin, V.; Marín-García, J. Mitochondrial oxidative metabolism and uncoupling proteins in the failing heart. Heart Fail. Rev. 2015, 20, 227–249. [Google Scholar] [CrossRef]
- Murray, A.J.; Anderson, R.E.; Watson, G.C.; Radda, G.K.; Clarke, K. Uncoupling proteins in human heart. Lancet 2004, 364, 1786–1788. [Google Scholar] [CrossRef]
- Wyss, M.; Kaddurah-Daouk, R. Creatine and creatinine metabolisms. Physiol. Rev. 2000, 80, 1107–1213. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, C.; Murakami, Y.; Gong, G.; Ishibashi, Y.; Prody, C.; Ochiai, K.; Bache, R.J.; Godinot, C.; Zhang, J. Mitochondrial ATPase and high-energy phosphates in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1319–H1326. [Google Scholar] [CrossRef] [PubMed]
- Conway, M.A.; Allis, J.; Ouwerkerk, R.; Niioka, T.; Rajagopalan, B.; Radda, G.K. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 1991, 338, 973–976. [Google Scholar] [CrossRef]
- Shen, W.; Asai, K.; Uechi, M.; Mathier, M.A.; Shannon, R.P.; Vatner, S.F.; Ingwall, J.S. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: A compensatory role for the parallel loss of creatine. Circulation 1999, 100, 2113–2118. [Google Scholar] [CrossRef]
- Starling, R.C.; Hammer, D.F.; Altschuld, R.A. Human myocardial ATP content and in vivo contractile function. Mol. Cell. Biochem. 1998, 180, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol. 2002, 40, 1267–1274. [Google Scholar] [CrossRef]
- Nascimben, L.; Ingwall, J.S.; Pauletto, P.; Friedrich, J.; Gwathmey, J.K.; Saks, V.; Pessina, A.C.; Allen, P.D. Creatine kinase system in failing and nonfailing human myocardium. Circulation 1996, 94, 1894–1901. [Google Scholar] [CrossRef]
- Kim, T.T.; Dyck, J.R. Is AMPK the savior of the failing heart? Trends Endocrinol. Metab. 2015, 26, 40–48. [Google Scholar] [CrossRef]
- Zaha, V.G.; Young, L.H. AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 2012, 111, 800–814. [Google Scholar] [CrossRef]
- Sasaki, H.; Asanuma, H.; Fujita, M.; Takahama, H.; Wakeno, M.; Ito, S.; Ogai, A.; Asakura, M.; Kim, J.; Minamino, T.; et al. Metformin prevents progression of heart failure in dogs: Role of amp-activated protein kinase. Circulation 2009, 119, 2568–2577. [Google Scholar] [CrossRef] [PubMed]
- Gundewar, S.; Calvert, J.W.; Jha, S.; Toedt-Pingel, I.; Ji, S.Y.; Nunez, D.; Ramachandran, A.; Anaya-Cisneros, M.; Tian, R.; Lefer, D.J. Activation of amp-activated protein kinase by metformin improves left ventricular function and survival in heart failure. Circ. Res. 2009, 104, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Coen, M.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. NMR-based metabolic profiling and metabonomic approaches to problems in molecular toxicology. Chem. Res. Toxicol. 2008, 21, 9–27. [Google Scholar] [CrossRef]
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The human metabolome database in 2013. Nucl. Acids Res. 2013, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Eisner, R.; Young, N.; Gautam, B.; Hau, D.D.; Psychogios, N.; Dong, E.; Bouatra, S.; et al. HMDB: A knowledgebase for the human metabolome. Nucl. Acids Res. 2009, 37, D603–D610. [Google Scholar] [CrossRef]
- Gika, H.G.; Theodoridis, G.A.; Wilson, I.D. Liquid chromatography and ultra-performance liquid chromatography-mass spectrometry fingerprinting of human urine: Sample stability under different handling and storage conditions for metabonomics studies. J. Chromatogr. A 2008, 1189, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.L.; Atherton, H.; Shockcor, J.; Atzori, L. Metabolomics as a tool for cardiac research. Nat. Rev. Cardiol. 2011, 8, 630–643. [Google Scholar] [CrossRef]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef]
- Andreadou, I.; Sigala, F.; Iliodromitis, E.K.; Papaefthimiou, M.; Sigalas, C.; Aligiannis, N.; Savvari, P.; Gorgoulis, V.; Papalabros, E.; Kremastinos, D.T. Acute doxorubicin cardiotoxicity is successfully treated with the phytochemical oleuropein through suppression of oxidative and nitrosative stress. J. Mol. Cell. Cardiol. 2007, 42, 549–558. [Google Scholar] [CrossRef]
- Andreadou, I.; Papaefthimiou, M.; Zira, A.; Constantinou, M.; Sigala, F.; Skaltsounis, A.L.; Tsantili-Kakoulidou, A.; Iliodromitis, E.K.; Kremastinos, D.T.; Mikros, E. Metabonomic identification of novel biomarkers in doxorubicin cardiotoxicity and protective effect of the natural antioxidant oleuropein. NMR Biomed. 2009, 22, 585–592. [Google Scholar] [CrossRef]
- Andreadou, I.; Mikros, E.; Ioannidis, K.; Sigala, F.; Naka, K.; Kostidis, S.; Farmakis, D.; Tenta, R.; Kavantzas, N.; Bibli, S.I.; et al. Oleuropein prevents doxorubicin-induced cardiomyopathy interfering with signaling molecules and cardiomyocyte metabolism. J. Mol. Cell. Cardiol. 2014, 69, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Lou, Z.; Liao, W.; Zhu, Z.; Dong, X.; Zhang, W.; Li, W.; Chai, Y. Potential biomarkers in mouse myocardium of doxorubicin-induced cardiomyopathy: A metabonomic method and its application. PLoS ONE 2011, 6, e27683. [Google Scholar] [CrossRef] [PubMed]
- Cong, W.; Liang, Q.; Li, L.; Shi, J.; Liu, Q.; Feng, Y.; Wang, Y.; Luo, G. Metabonomic study on the cumulative cardiotoxicity of a pirarubicin liposome powder. Talanta 2012, 89, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ju, L.; Hou, Z.; Deng, H.; Zhang, Z.; Wang, L.; Yang, Z.; Yin, J.; Zhang, Y. Screening, verification, and optimization of biomarkers for early prediction of cardiotoxicity based on metabolomics. J. Proteome Res. 2015, 14, 2437–2445. [Google Scholar] [CrossRef] [PubMed]
- Schnackenberg, L.K.; Pence, L.; Vijay, V.; Moland, C.L.; George, N.; Cao, Z.; Yu, L.R.; Fuscoe, J.C.; Beger, R.D.; Desai, V.G. Early metabolomics changes in heart and plasma during chronic doxorubicin treatment in B6C3F1 mice. J. Appl. Toxicol. 2016, 36, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Xie, J.; Guo, X.; Ju, L.; Li, Y.; Zhang, Y. Plasma metabolic profiling analysis of cyclophosphamide-induced cardiotoxicity using metabolomics coupled with UPLC/Q-TOF-MS and ROC curve. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1033, 428–435. [Google Scholar] [CrossRef]
- Chaudhari, U.; Ellis, J.K.; Wagh, V.; Nemade, H.; Hescheler, J.; Keun, H.C.; Sachinidis, A. Metabolite signatures of doxorubicin induced toxicity in human induced pluripotent stem cell-derived cardiomyocytes. Amino Acids 2017, 49, 1955–1963. [Google Scholar] [CrossRef]
- QuanJun, Y.; GenJin, Y.; LiLi, W.; YongLong, H.; Yan, H.; Jie, L.; JinLu, H.; Jin, L.; Run, G.; Cheng, G. Protective effects of dexrazoxane against doxorubicin-induced cardiotoxicity: A metabolomic study. PLoS ONE 2017, 12, e0169567. [Google Scholar] [CrossRef]
- Jensen, B.C.; Parry, T.L.; Huang, W.; Beak, J.Y.; Ilaiwy, A.; Bain, J.R.; Newgard, C.B.; Muehlbauer, M.J.; Patterson, C.; Johnson, G.L.; et al. Effects of the kinase inhibitor sorafenib on heart, muscle, liver and plasma metabolism in vivo using non-targeted metabolomics analysis. Br. J. Pharmacol. 2017, 174, 4797–4811. [Google Scholar] [CrossRef]
- Jensen, B.C.; Parry, T.L.; Huang, W.; Ilaiwy, A.; Bain, J.R.; Muehlbauer, M.J.; O’Neal, S.K.; Patterson, C.; Johnson, G.L.; Willis, M.S. Non-Targeted metabolomics analysis of the effects of tyrosine kinase inhibitors sunitinib and erlotinib on heart, muscle, liver and serum metabolism in vivo. Metabolites 2017, 7, 31. [Google Scholar] [CrossRef]
- Yoon, C.S.; Kim, H.K.; Mishchenko, N.P.; Vasileva, E.A.; Fedoreyev, S.A.; Stonik, V.A.; Han, J. Spinochrome D attenuates doxorubicin-induced cardiomyocyte death via improving glutathione metabolism and attenuating oxidative stress. Mar. Drugs 2018, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Gramatyka, M.; Skorupa, A.; Sokół, M. Nuclear magnetic resonance spectroscopy reveals metabolic changes in living cardiomyocytes after low doses of ionizing radiation. Acta Biochim. Pol. 2018, 65, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Madonna, R.; Cadeddu, C.; Deidda, M.; Giricz, Z.; Madeddu, C.; Mele, D.; Monte, I.; Novo, G.; Pagliaro, P.; Pepe, A.; et al. Cardioprotection by gene therapy: A review paper on behalf of the working group on drug cardiotoxicity and cardioprotection of the Italian Society of Cardiology. Int. J. Cardiol. 2015, 191, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Tocchetti, C.G.; Cadeddu, C.; Di Lisi, D.; Femminò, S.; Madonna, R.; Mele, D.; Monte, I.; Novo, G.; Penna, C.; Pepe, A.; et al. From Molecular Mechanisms to Clinical Management of Antineoplastic Drug-Induced Cardiovascular Toxicity: A Translational Overview. Antioxid. Redox Signal. 2019, 30, 2110–2153. [Google Scholar] [CrossRef] [PubMed]
- Cadeddu, C.; Mercurio, V.; Spallarossa, P.; Nodari, S.; Triggiani, M.; Monte, I.; Piras, R.; Madonna, R.; Pagliaro, P.; Tocchetti, C.G.; et al. Preventing antiblastic drug-related cardiomyopathy: Old and new therapeutic strategies. J. Cardiovasc. Med. (Hagerstown) 2016, 17 (Suppl. S1), S64–S75. [Google Scholar] [CrossRef] [PubMed]



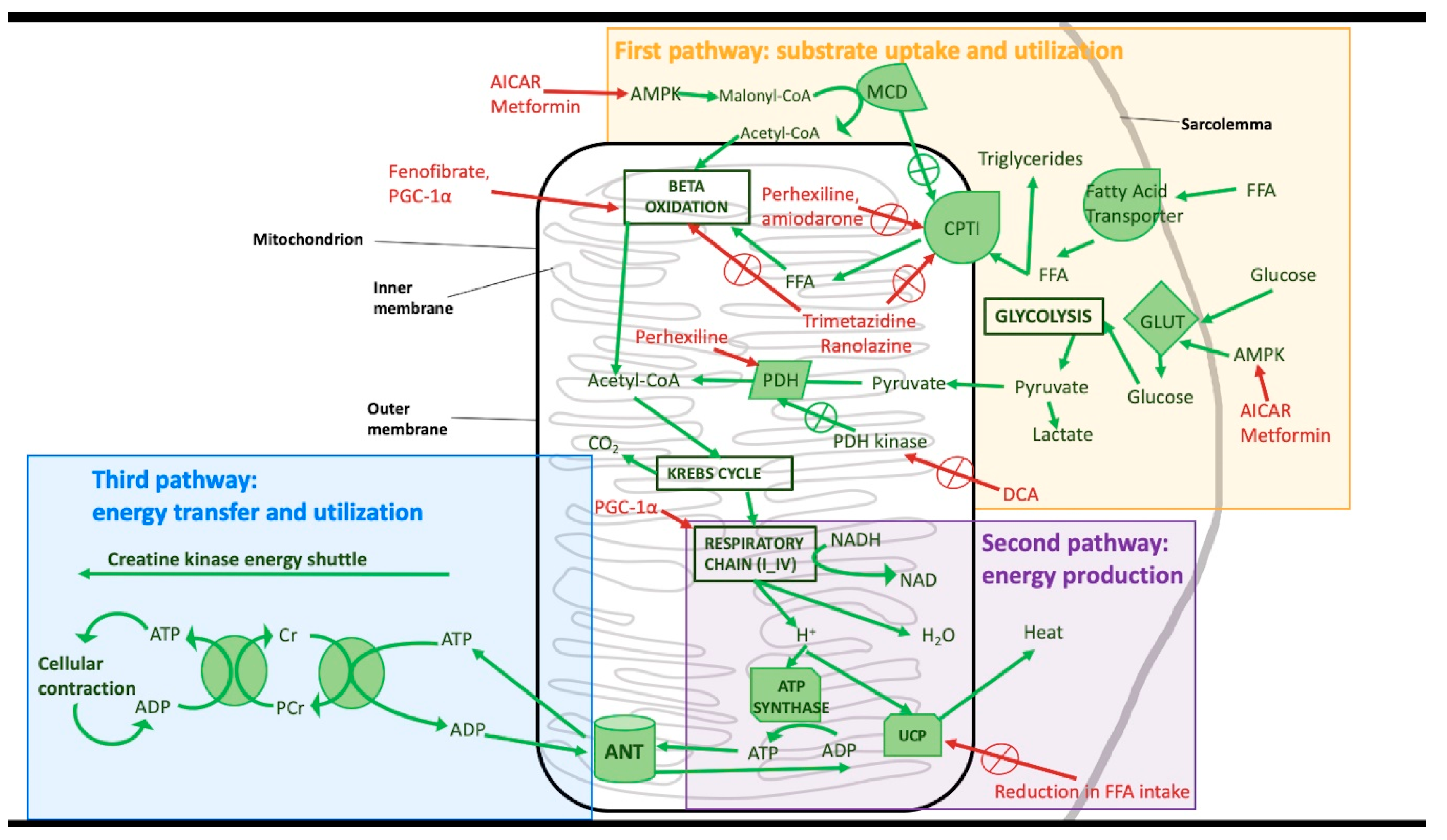{kind=link}
{kind=link}
| Technique | Cost | Throughput | Advantages | Disadvantages |
|---|---|---|---|---|
| High-resolution NMR spectroscopy (NMR) | Low per sample | ~10 min | Simultaneous detection of many different compounds, such as carbohydrates, amino acids, organic and fatty Acids, amines and lipids without any initial sample pre-treatment Non-destructive technique Good libraries of spectra Easy to process | Poor sensitivity Co-resonances for 1D-NMR spectroscopy 2D-NMR spectroscopy is time-consuming |
| In vivo NMR spectroscopy | High | ~30 min with preparation time | Possibility of observing the metabolism of the working heart Imaging can map metabolite distributions | Very poor sensitivity (hyperpolarization or higher field strengths could improve it) |
| High-resolution magic-angle-spinning NMR (HR-MAS-NMR) | Low | ~15 min | Possibility of monitoring the cellular environment (e.g., compartmentation) in intact tissue Tissue can be chilled to reduce post-mortem effects | Tissue cannot be perfused, so its viability is limited |
| Direct-infusion MS | Low | 3–4 min | Has been used to profile both aqueous and lipophilic metabolites in various studies Minimal carry-over, as no chromatography involved Good reproducibility Simple to optimise | Ion suppression can be a substantial problem; identification can require chromatography, e.g., for isobaric species; metabolite identification is a significant challenge and requires mass spectometry acquisitions; semiquantitative at best |
| GC–MS | Low-medium | 20–30 min for FA 30–45 min for aqueous metabolites | Chromatography is robust and reproducible Metabolite identification is aided by the adoption of standard ionisation parameters in electron impact Can be quantitative | Metabolites need derivatisation, and not all metabolites are suitable for derivatisation |
| LC–MS | Medium | ~15–30 min | Chromatography reduces the effect of ion suppression and can separate isobaric species Suitable for measuring intact lipids, dipeptides, tripeptides, and other macromolecules | Chromatography can drift during a sample run, which makes data processing difficult Metabolite identification is a major challenge |
| Triple quadrupole (targeted) MS | Medium to high | 15 min per chromatography run ~60 min for more comprehensive screens | Highly sensitive Highly quantitative Targeted Results readily transferable because concentrations can be measured | Targeted, so the discovery of novel biomarkers is unlikely Time-consuming to set up quantitative assays |
| Reference | Species | Technique | Biofluid/Tissue | Metabolites/Metabolism Discrimination |
|---|---|---|---|---|
| Andreadu et al., 2009 [50] | Wistar rats | NMR | Aqueous myocardial extracts | Increased levels of acetate and succinate, decreased levels of branched-chain amino acids |
| Andreadu et al., 2014 [51] | Wistar rats | NMR | Aqueous myocardial extracts | Perturbations of energy metabolism |
| Tan et al., 2011 [52] | ICR mice | GC–MS | Myocardial tissue | Increased levels of l-alanine, phosphate, glycine, succinate, malate, proline, threonic acid, glutamine, phenylalanine, dihydroxyacetonephosphate (DHAP), glycerol-3-phosphate (G-3-P), fructose, glucose, stearic acid, myo-inositol and cholesterol; decreased levels of lactate, β-hydroxybutyric acid, l-valine, isoleucine, threonine, citrate, linoleic acid, arachidonic acid |
| Cong et al., 2012 [53] | Sprague-Dawley rats | UPLC–TOF-MS | Urine | Metabolites involved in metabolic process related to myocardial energy metabolism: tricarboxylic acid cycle (citrate), glycolysis (lactate), pentose phosphate pathway (d-gluconate-1-phosphate) and amino acid metabolism (N-acetylglutamine and N-acetyl-dl-tryptophan) |
| Li et al., 2015 [54] | Wistar rats | UPLC–Q-TOF-MS | Plasma | l-carnitine, 19-hydroxydioxycortic acid, LPC (14:0) and LPC (20:2) |
| Schnackenberg et al., 2016 [55] | B6C3F1 mice | GC-MS, NMR | Heart tissue, Plasma | Myocardial specimens: altered levels of 18 amino acids and acetylornithine, kynurenine, putrescine and serotonin, decreased levels of 5 acylcarnitines. Plasma samples: altered levels of 16 amino acids and acetylornithine and hydroxyproline, increased levels of 16 acylcarnitines |
| Yin et al., 2016 [56] | Wistar rats | UPLC–Q-TOF-MS | Plasma | l-carnitine, proline, 19-hydroxydeoxycorticosterone, phuyoshingosine, cholic acid, LPC (14:0), LPC (18:3), LPC (16:1), LPE (18:2), LPC (22:5), LPC (22:6), linoleic acid, LPC (22:4), LPC (20:2), LPE (18:0), LPC (20:3) |
| Chaudhari et al., 2017 [57] | Human induced pluripotent stem cell-derived cardiomyocytes | NMR | Culture medium | Reduction in the utilisation of pyruvate and acetate, and accumulation of formate |
| QuanJun et al., 2017 [58] | BALB/c mice | NMR | Serum | DOX administration: increase in 5-hydroxylisine, 2-hydroxybutyrate, 2-oxoglutarate, 3-hydroxybutyrate decrease in glucose, glutamate, cysteine, acetone, methionine, asparate, isoleucine and glycylproline. DZR treatment: increase in lactate, 3-hydroxybutyrate, glutamate, alanine; decrease in glucose, trimethylamine N-oxide and carnosine levels |
| Jensen et al., 2017 [59] | FVB/N mice | GC–MS | Plasma and heart, skeletal muscle and liver tissues | Significant alterations in 11 metabolites, including markedly altered taurine/hypotaurine metabolism: glutamine, ethanolamine, stearamide, taurine, O-phosphocolamine, hypotaurine, myo-inosithol-2-phosphate, dehydroalanine, adenosine-5-monophosphate, glycerol-1-phosphate |
| Jensen et al., 2017 [60] | FVB/N mice | GC-MS | Serum and heart, skeletal muscle and liver tissues | Significantly lower heart and skeletal muscle levels of long chain omega-3 fatty acids docosahexaenoic acid (DHA), arachidonic acid (AA)/eicosapentaenoic acid (EPA) and increased serum O-phosphocholine phospholipid |
| Yoon et al., 2019 [61] | Human cardiomyocytes | NMR | Cardiomyocites | Decrease of acetate, glutamine, serine, uracil, glycerol; increase of glutamate, isoleucine, O-phosphocholine, taurine, myo-inositol, glutathione, sn-glycero-3-phosphocholine |
| Gramatyka et al., 2018 [62] | Human cardiomyocytes | HR-MAS NMR (High-Resolution Magic-Angle-Spinning Nuclear Magnetic Resonance) | Cardiomyocites | Lipids, threonine, glycine, glycerophosphocholine, choline, valine, isoleucine, glutamate; reduced glutathione and taurine metabolism |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deidda, M.; Mercurio, V.; Cuomo, A.; Noto, A.; Mercuro, G.; Cadeddu Dessalvi, C. Metabolomic Perspectives in Antiblastic Cardiotoxicity and Cardioprotection. Int. J. Mol. Sci. 2019, 20, 4928. https://doi.org/10.3390/ijms20194928
Deidda M, Mercurio V, Cuomo A, Noto A, Mercuro G, Cadeddu Dessalvi C. Metabolomic Perspectives in Antiblastic Cardiotoxicity and Cardioprotection. International Journal of Molecular Sciences. 2019; 20(19):4928. https://doi.org/10.3390/ijms20194928
Chicago/Turabian StyleDeidda, Martino, Valentina Mercurio, Alessandra Cuomo, Antonio Noto, Giuseppe Mercuro, and Christian Cadeddu Dessalvi. 2019. "Metabolomic Perspectives in Antiblastic Cardiotoxicity and Cardioprotection" International Journal of Molecular Sciences 20, no. 19: 4928. https://doi.org/10.3390/ijms20194928
APA StyleDeidda, M., Mercurio, V., Cuomo, A., Noto, A., Mercuro, G., & Cadeddu Dessalvi, C. (2019). Metabolomic Perspectives in Antiblastic Cardiotoxicity and Cardioprotection. International Journal of Molecular Sciences, 20(19), 4928. https://doi.org/10.3390/ijms20194928