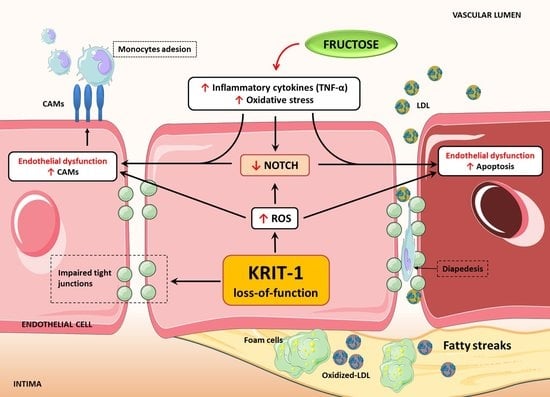
KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction


 , , , ,
, , , , 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
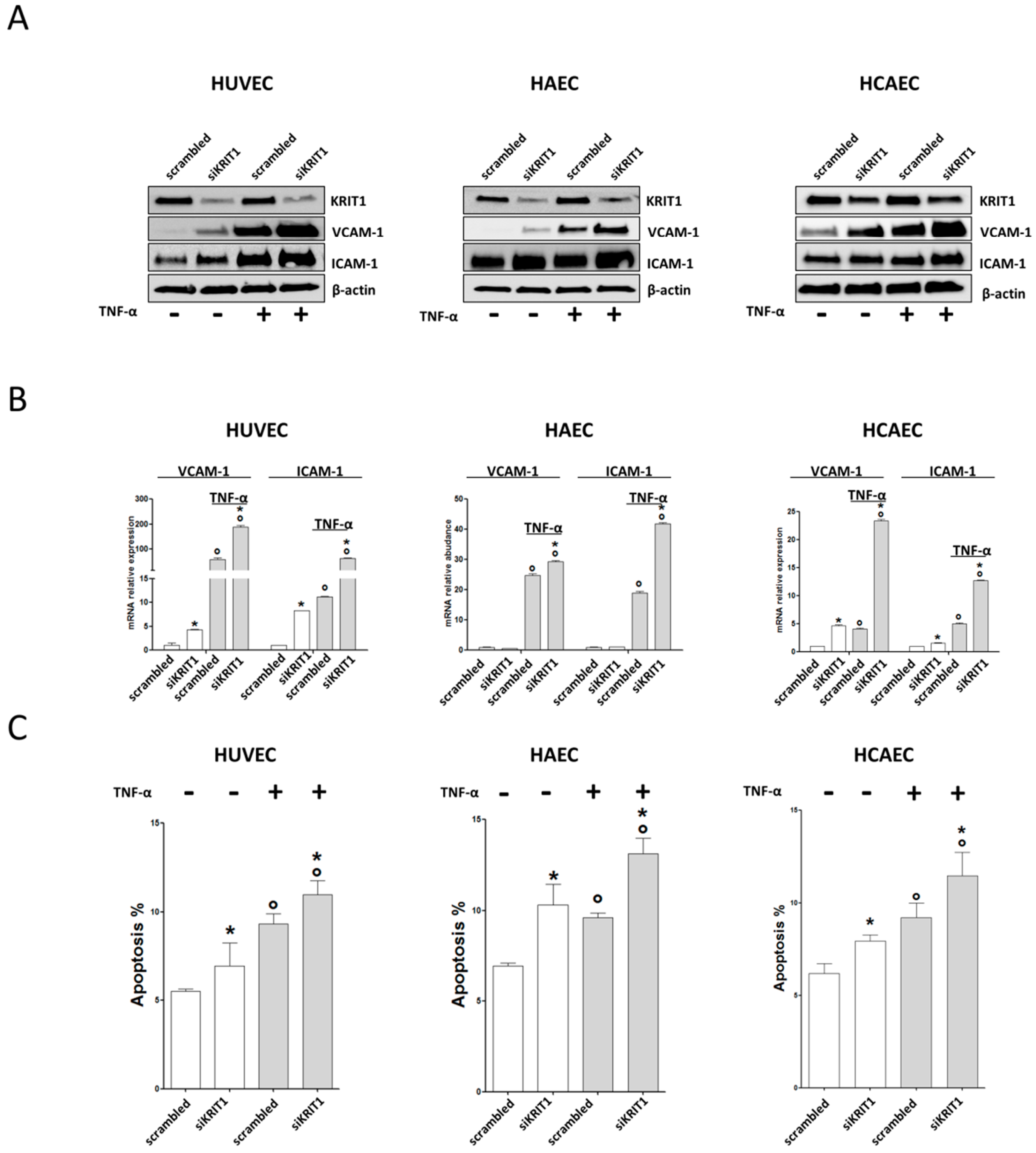
2.1. KRIT1 Downregulation Causes Increased Susceptibility to Inflammation-Induced Endothelial Dysfunction
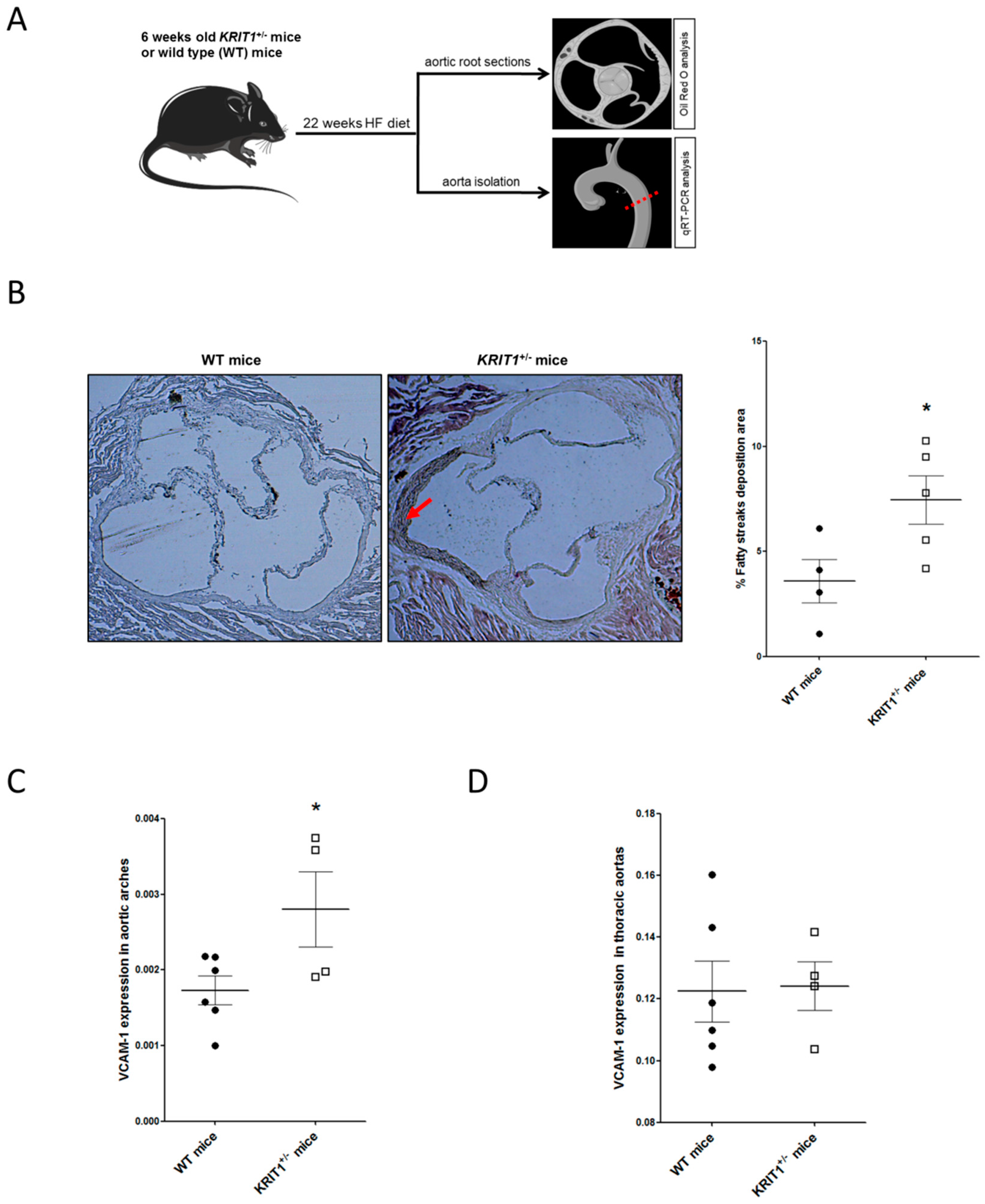
2.2. KRIT1+/− Mice Show an Increased Susceptibility to High-Fructose Diet-Induced Aortic Endothelial Dysfunction
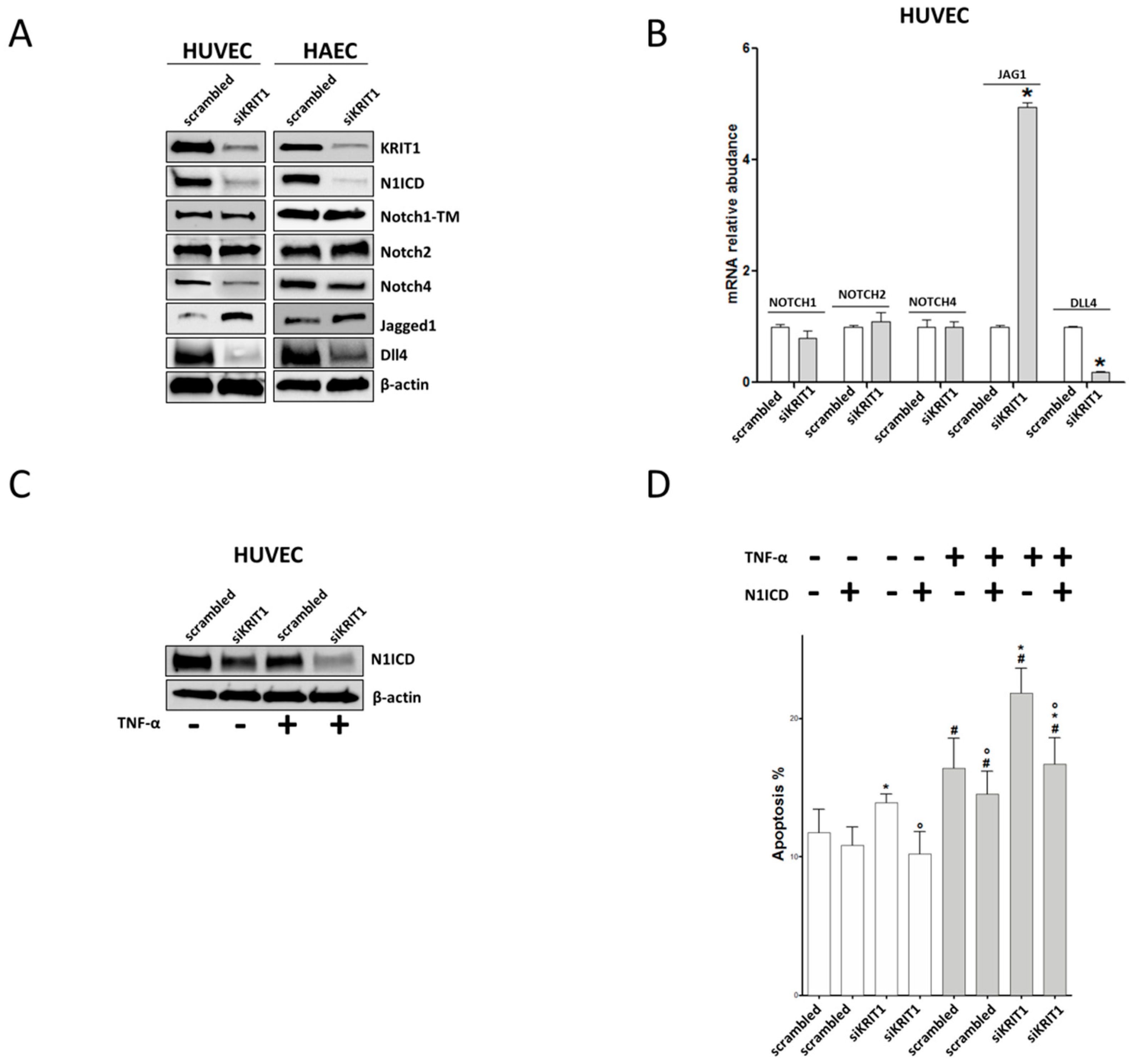
2.3. KRIT1 Downregulation Leads to Endothelial Dysfunction through Inhibition of Notch1
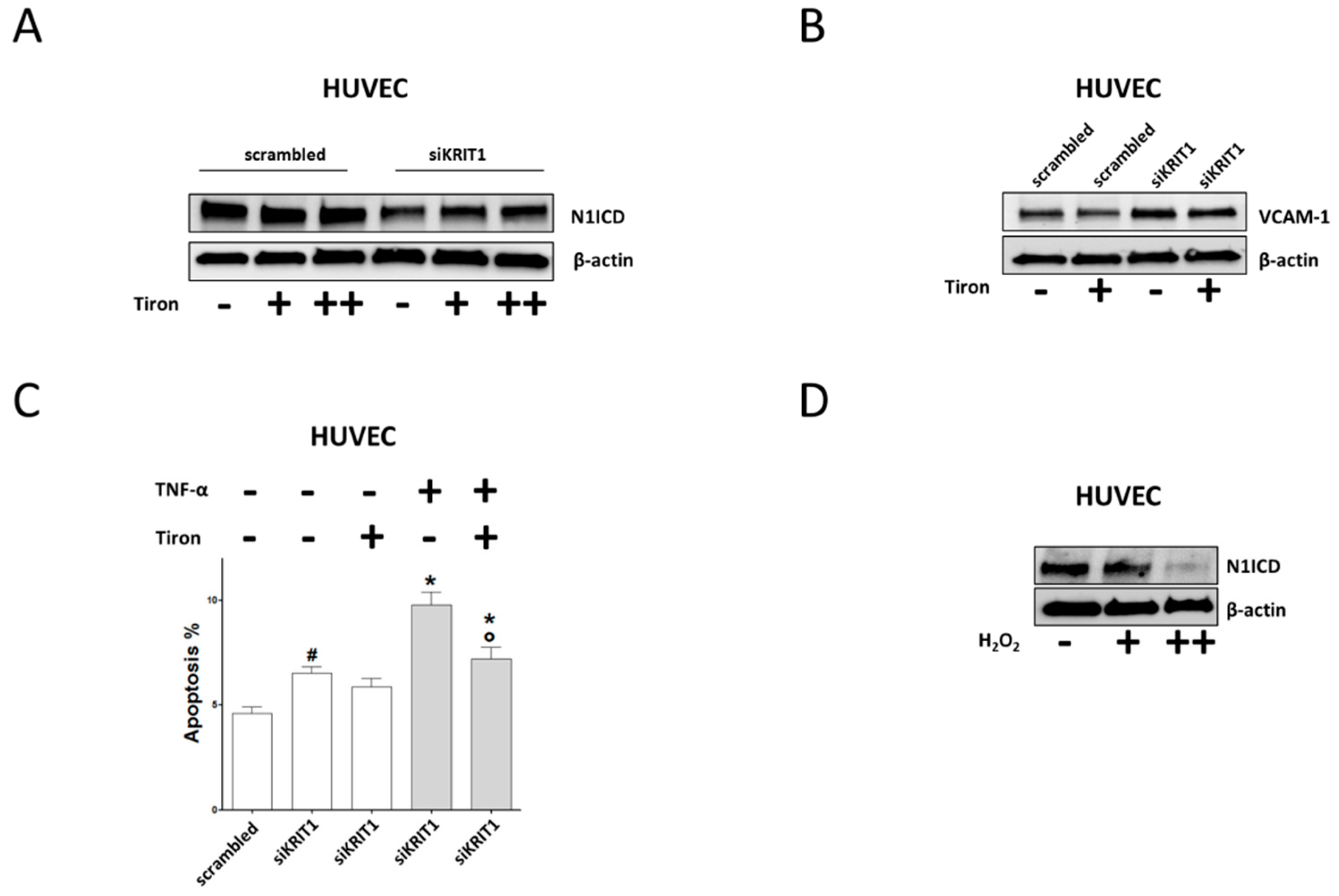
2.4. Downregulation of Notch Signaling and Upregulation of VCAM-1 and Apoptosis are Redox-Dependent Effects of KRIT1 Loss-of-Function
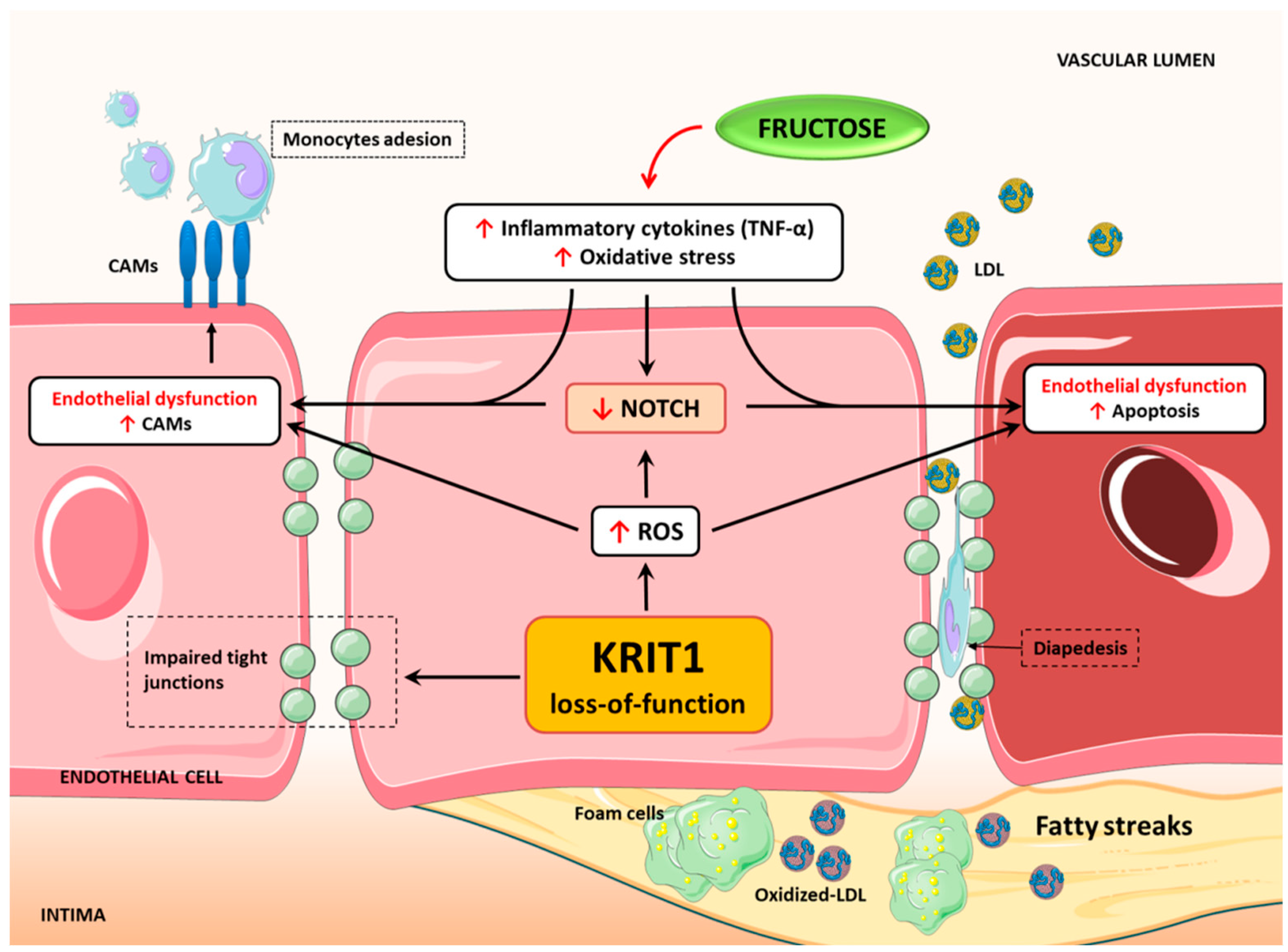
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Animal Models
4.3. Ethics Statement
4.4. Fatty Streak Analysis in Section of Aortic Root
4.5. Cell Transfection
4.6. Real-Time PCR
4.7. Western Blotting
4.8. Apoptosis Detection
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| KRIT1 | Krev interaction trapped protein 1 |
| CCM | Cerebral Cavernous Malformation |
| HUVEC | human umbilical vein endothelial cell |
| HAEC | human aortic endothelial cell |
| HCAEC | human coronary artery endothelial cell |
| ED | endothelial dysfunction |
| N1ICD | Notch1 intracellular domain |
References
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Vaccarezza, M.; Balla, C.; Rizzo, P. Atherosclerosis as an inflammatory disease: Doubts? No more. Int. J. Cardiol. Heart Vasc. 2018, 19, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Retta, S.F.; Glading, A.J. Oxidative stress and inflammation in cerebral cavernous malformation disease pathogenesis: Two sides of the same coin. Int. J. Biochem. Cell Biol. 2016, 81, 254–270. [Google Scholar] [CrossRef] [PubMed]
- Guazzi, P.; Goitre, L.; Ferro, E.; Cutano, V.; Martino, C.; Trabalzini, L.; Retta, S.F. Identification of the Kelch family protein Nd1-L as a novel molecular interactor of KRIT1. PLoS ONE 2012, 7, e44705. [Google Scholar] [CrossRef]
- Draheim, K.M.; Fisher, O.S.; Boggon, T.J.; Calderwood, D.A. Cerebral cavernous malformation proteins at a glance. J. Cell Sci. 2014, 127, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Fisher, O.S.; Boggon, T.J. Signaling pathways and the cerebral cavernous malformations proteins: Lessons from structural biology. Cell Mol. Life Sci. 2014, 71, 1881–1892. [Google Scholar] [CrossRef]
- Glading, A.; Han, J.; Stockton, R.A.; Ginsberg, M.H. KRIT-1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. J. Cell Biol. 2007, 179, 247–254. [Google Scholar] [CrossRef]
- Faurobert, E.; Rome, C.; Lisowska, J.; Manet-Dupé, S.; Boulday, G.; Malbouyres, M.; Balland, M.; Bouin, A.P.; Kéramidas, M.; Bouvard, D.; et al. CCM1-ICAP-1 complex controls β1 integrin-dependent endothelial contractility and fibronectin remodeling. J. Cell Biol. 2013, 202, 545–561. [Google Scholar] [CrossRef]
- Liu, W.; Draheim, K.M.; Zhang, R.; Calderwood, D.A.; Boggon, T.J. Mechanism for KRIT1 release of ICAP1-mediated suppression of integrin activation. Mol. Cell 2013, 49, 719–729. [Google Scholar] [CrossRef]
- Macek Jilkova, Z.; Lisowska, J.; Manet, S.; Verdier, C.; Deplano, V.; Geindreau, C.; Faurobert, E.; Albigès-Rizo, C.; Duperray, A. CCM proteins control endothelial β1 integrin dependent response to shear stress. Biol. Open 2014, 3, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.J.; Chan, A.C.; Navankasattusas, S.; Koh, W.; London, N.R.; Ling, J.; Mayo, A.H.; Drakos, S.G.; Jones, C.A.; Zhu, W.; et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009, 15, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896. [Google Scholar] [CrossRef] [PubMed]
- Schulz, G.B.; Wieland, E.; Wüstehube-Lausch, J.; Boulday, G.; Moll, I.; Tournier-Lasserve, E.; Fischer, A. Cerebral Cavernous Malformation-1 Protein Controls DLL4-Notch3 Signaling Between the Endothelium and Pericytes. Stroke 2015, 46, 1337–1343. [Google Scholar] [CrossRef]
- Wüstehube, J.; Bartol, A.; Liebler, S.S.; Brütsch, R.; Zhu, Y.; Felbor, U.; Sure, U.; Augustin, H.G.; Fischer, A. Cerebral cavernous malformation protein CCM1 inhibits sprouting angiogenesis by activating DELTA-NOTCH signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 12640–12645. [Google Scholar] [CrossRef] [PubMed]
- You, C.; Sandalcioglu, I.E.; Dammann, P.; Felbor, U.; Sure, U.; Zhu, Y. Loss of CCM3 impairs DLL4-Notch signalling: Implication in endothelial angiogenesis and in inherited cerebral cavernous malformations. J. Cell Mol. Med. 2013, 17, 407–418. [Google Scholar] [CrossRef]
- DiStefano, P.V.; Kuebel, J.M.; Sarelius, I.H.; Glading, A.J. KRIT1 protein depletion modifies endothelial cell behavior via increased vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 2014, 289, 33054–33065. [Google Scholar] [CrossRef]
- Renz, M.; Otten, C.; Faurobert, E.; Rudolph, F.; Zhu, Y.; Boulday, G.; Duchene, J.; Mickoleit, M.; Dietrich, A.C.; Ramspacher, C.; et al. Regulation of β1 integrin-Klf2-mediated angiogenesis by CCM proteins. Dev. Cell 2015, 32, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tang, A.T.; Wong, W.Y.; Bamezai, S.; Goddard, L.M.; Shenkar, R.; Zhou, S.; Yang, J.; Wright, A.C.; Foley, M.; et al. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature 2016, 532, 122–126. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptive homeostasis that sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef]
- Goitre, L.; Balzac, F.; Degani, S.; Degan, P.; Marchi, S.; Pinton, P.; Retta, S.F. KRIT1 regulates the homeostasis of intracellular reactive oxygen species. PLoS ONE 2010, 5, e11786. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; De Luca, E.; Braggion, S.; Trapani, E.; Guglielmotto, M.; Biasi, F.; Forni, M.; Moglia, A.; Trabalzini, L.; Retta, S.F. KRIT1 loss of function causes a ROS-dependent upregulation of c-Jun. Free Radic. Biol. Med. 2014, 68, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Goitre, L.; DiStefano, P.V.; Moglia, A.; Nobiletti, N.; Baldini, E.; Trabalzini, L.; Keubel, J.; Trapani, E.; Shuvaev, V.V.; Muzykantov, V.R.; et al. Up-regulation of NADPH oxidase-mediated redox signaling contributes to the loss of barrier function in KRIT1 deficient endothelium. Sci. Rep. 2017, 7, 8296. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Corricelli, M.; Trapani, E.; Bravi, L.; Pittaro, A.; Delle Monache, S.; Ferroni, L.; Patergnani, S.; Missiroli, S.; Goitre, L.; et al. Defective autophagy is a key feature of cerebral cavernous malformations. EMBO Mol. Med. 2015, 7, 1403–1417. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Trapani, E.; Corricelli, M.; Goitre, L.; Pinton, P.; Retta, S.F. Beyond multiple mechanisms and a unique drug: Defective autophagy as pivotal player in cerebral cavernous malformation pathogenesis and implications for targeted therapies. Rare Dis. 2016, 4, e1142640. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Fornelli, C.; Retta, F.; Cassoni, P.; Talesa, V.N.; Retta, S.F. Data in support of sustained upregulation of adaptive redox homeostasis mechanisms caused by KRIT1 loss-of-function. Data Brief 2018, 16, 929–938. [Google Scholar] [CrossRef]
- Marchi, S.; Retta, S.F.; Pinton, P. Cellular processes underlying cerebral cavernous malformations: Autophagy as another point of view. Autophagy 2016, 12, 424–425. [Google Scholar] [CrossRef] [PubMed]
- Cianfruglia, L.; Perrelli, A.; Fornelli, C.; Magini, A.; Gorbi, S.; Salzano, A.M.; Antognelli, C.; Retta, F.; Benedetti, V.; Cassoni, P.; et al. KRIT1 Loss-Of-Function Associated with Cerebral Cavernous Malformation Disease Leads to Enhanced. Antioxidants 2019, 8, 27. [Google Scholar] [CrossRef]
- Corr, M.; Lerman, I.; Keubel, J.M.; Ronacher, L.; Misra, R.; Lund, F.; Sarelius, I.H.; Glading, A.J. Decreased Krev interaction-trapped 1 expression leads to increased vascular permeability and modifies inflammatory responses in vivo. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2702–2710. [Google Scholar] [CrossRef]
- Gibson, C.C.; Zhu, W.; Davis, C.T.; Bowman-Kirigin, J.A.; Chan, A.C.; Ling, J.; Walker, A.E.; Goitre, L.; Delle Monache, S.; Retta, S.F.; et al. Strategy for identifying repurposed drugs for the treatment of cerebral cavernous malformation. Circulation 2015, 131, 289–299. [Google Scholar] [CrossRef]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Orso, F.; Balzac, F.; Marino, M.; Lembo, A.; Retta, S.F.; Taverna, D. miR-21 coordinates tumor growth and modulates KRIT1 levels. Biochem. Biophys. Res. Commun. 2013, 438, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Qu, G.; Han, C.; Wang, Y.; Sun, T.; Li, F.; Wang, J.; Luo, S. MiR-34a, miR-21 and miR-23a as potential biomarkers for coronary artery disease: A pilot microarray study and confirmation in a 32 patient cohort. Exp. Mol. Med. 2015, 47, e138. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.J.; Plummer, N.W.; Adams, J.A.; Marchuk, D.A.; Li, D.Y. Ccm1 is required for arterial morphogenesis: Implications for the etiology of human cavernous malformations. Development 2004, 131, 1437–1448. [Google Scholar] [CrossRef]
- Trapani, E.; Retta, S.F. Cerebral cavernous malformation (CCM) disease: From monogenic forms to genetic susceptibility factors. J. Neurosurg. Sci. 2015, 59, 201–209. [Google Scholar]
- Choquet, H.; Trapani, E.; Goitre, L.; Trabalzini, L.; Akers, A.; Fontanella, M.; Hart, B.L.; Morrison, L.A.; Pawlikowska, L.; Kim, H.; et al. Cytochrome P450 and matrix metalloproteinase genetic modifiers of disease severity in Cerebral Cavernous Malformation type 1. Free Radic. Biol. Med. 2016, 92, 100–109. [Google Scholar] [CrossRef]
- Tang, A.T.; Choi, J.P.; Kotzin, J.J.; Yang, Y.; Hong, C.C.; Hobson, N.; Girard, R.; Zeineddine, H.A.; Lightle, R.; Moore, T.; et al. Endothelial TLR4 and the microbiome drive cerebral cavernous malformations. Nature 2017, 545, 305–310. [Google Scholar] [CrossRef]
- Tappy, L.; Lê, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef]
- Mackay, F.; Loetscher, H.; Stueber, D.; Gehr, G.; Lesslauer, W. Tumor necrosis factor alpha (TNF-alpha)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993, 177, 1277–1286. [Google Scholar] [CrossRef]
- Fortini, F.; Vieceli Dalla Sega, F.; Caliceti, C.; Aquila, G.; Pannella, M.; Pannuti, A.; Miele, L.; Ferrari, R.; Rizzo, P. Estrogen receptor β-dependent Notch1 activation protects vascular endothelium against tumor necrosis factor α (TNFα)-induced apoptosis. J. Biol. Chem. 2017, 292, 18178–18191. [Google Scholar] [CrossRef] [PubMed]
- Endemann, D.H.; Schiffrin, E.L. Endothelial dysfunction. J. Am. Soc. Nephrol. 2004, 15, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.V.; Kiat, H. The mechanisms underlying fructose-induced hypertension: A review. J. Hypertens. 2015, 33, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Ahn, H.; Park, Y.K. High Dietary Fructose Intake on Cardiovascular Disease Related Parameters in Growing Rats. Nutrients 2016, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.M.; Jiao, R.Q.; Kong, L.D. High Dietary Fructose: Direct or Indirect Dangerous Factors Disturbing Tissue and Organ Functions. Nutrients 2017, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Tokita, Y.; Hirayama, Y.; Sekikawa, A.; Kotake, H.; Toyota, T.; Miyazawa, T.; Sawai, T.; Oikawa, S. Fructose ingestion enhances atherosclerosis and deposition of advanced glycated end-products in cholesterol-fed rabbits. J. Atheroscler. Thromb. 2005, 12, 260–267. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kolderup, A.; Svihus, B. Fructose Metabolism and Relation to Atherosclerosis, Type 2 Diabetes, and Obesity. J. Nutr. Metab. 2015, 2015, 823081. [Google Scholar] [CrossRef]
- Collotta, D.; Lucarini, L.; Chiazza, F.; Cento, A.S.; Durante, M.; Sgambellone, S.; Chini, J.; Baratta, F.; Aragno, M.; Mastrocola, R.; et al. Reduced Susceptibility to Sugar-Induced Metabolic Derangements and Impairments of Myocardial Redox Signaling in Mice Chronically Fed with D-Tagatose when Compared to Fructose. Oxid. Med. Cell Longev. 2018, 2018, 5042428. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Nigro, D.; Chiazza, F.; Medana, C.; Dal Bello, F.; Boccuzzi, G.; Collino, M.; Aragno, M. Fructose-derived advanced glycation end-products drive lipogenesis and skeletal muscle reprogramming via SREBP-1c dysregulation in mice. Free Radic. Biol. Med. 2016, 91, 224–235. [Google Scholar] [CrossRef]
- Mastrocola, R.; Nigro, D.; Cento, A.S.; Chiazza, F.; Collino, M.; Aragno, M. High-fructose intake as risk factor for neurodegeneration: Key role for carboxy methyllysine accumulation in mice hippocampal neurons. Neurobiol. Dis. 2016, 89, 65–75. [Google Scholar] [CrossRef]
- Zibara, K.; Chignier, E.; Covacho, C.; Poston, R.; Canard, G.; Hardy, P.; McGregor, J. Modulation of expression of endothelial intercellular adhesion molecule-1, platelet-endothelial cell adhesion molecule-1, and vascular cell adhesion molecule-1 in aortic arch lesions of apolipoprotein E-deficient compared with wild-type mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Haidari, M.; Ali, M.; Gangehei, L.; Chen, M.; Zhang, W.; Cybulsky, M.I. Increased oxidative stress in atherosclerosis-predisposed regions of the mouse aorta. Life Sci. 2010, 87, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Iiyama, K.; Hajra, L.; Iiyama, M.; Li, H.; DiChiara, M.; Medoff, B.D.; Cybulsky, M.I. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ. Res. 1999, 85, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, R.; Tonar, Z.; Bartova, J.; Nedorost, L.; Rossman, P.; Poledne, R.; Schwarzer, M.; Tlaskalova-Hogenova, H. Absence of microbiota (germ-free conditions) accelerates the atherosclerosis in ApoE-deficient mice fed standard low cholesterol diet. J. Atheroscler. Thromb. 2010, 17, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Merat, S.; Casanada, F.; Sutphin, M.; Palinski, W.; Reaven, P.D. Western-type diets induce insulin resistance and hyperinsulinemia in LDL receptor-deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fructose-rich diet. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, P.; Miele, L.; Ferrari, R. The Notch pathway: A crossroad between the life and death of the endothelium. Eur. Heart J. 2013, 34, 2504–2509. [Google Scholar] [CrossRef]
- Vieceli Dalla Sega, F.; Aquila, G.; Fortini, F.; Vaccarezza, M.; Secchiero, P.; Rizzo, P.; Campo, G. Context-dependent function of ROS in the vascular endothelium: The role of the Notch pathway and shear stress. Biofactors 2017, 43, 475–485. [Google Scholar] [CrossRef]
- Lin, Q.Q.; Zhao, J.; Zheng, C.G.; Chun, J. Roles of notch signaling pathway and endothelial-mesenchymal transition in vascular endothelial dysfunction and atherosclerosis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 6485–6491. [Google Scholar] [CrossRef]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef]
- Gridley, T. Notch signaling in the vasculature. Curr. Top. Dev. Biol. 2010, 92, 277–309. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Hellström, M.; Phng, L.K.; Hofmann, J.J.; Wallgard, E.; Coultas, L.; Lindblom, P.; Alva, J.; Nilsson, A.K.; Karlsson, L.; Gaiano, N.; et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 2007, 445, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Osipo, C.; Patel, P.; Rizzo, P.; Clementz, A.G.; Hao, L.; Golde, T.E.; Miele, L. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a γ-secretase inhibitor. Oncogene 2008, 27, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Bouloumié, A.; Iruela-Arispe, M.L. Notch, lipids, and endothelial cells. Curr. Opin. Lipidol. 2016, 27, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Briot, A.; Civelek, M.; Seki, A.; Hoi, K.; Mack, J.J.; Lee, S.D.; Kim, J.; Hong, C.; Yu, J.; Fishbein, G.A.; et al. Endothelial NOTCH1 is suppressed by circulating lipids and antagonizes inflammation during atherosclerosis. J. Exp. Med. 2015, 212, 2147–2163. [Google Scholar] [CrossRef]
- Quillard, T.; Coupel, S.; Coulon, F.; Fitau, J.; Chatelais, M.; Cuturi, M.C.; Chiffoleau, E.; Charreau, B. Impaired Notch4 activity elicits endothelial cell activation and apoptosis: Implication for transplant arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Devalliere, J.; Chatelais, M.; Coulon, F.; Séveno, C.; Romagnoli, M.; Barillé Nion, S.; Charreau, B. Notch2 signaling sensitizes endothelial cells to apoptosis by negatively regulating the key protective molecule survivin. PLoS ONE 2009, 4, e8244. [Google Scholar] [CrossRef]
- Kar, S.; Baisantry, A.; Nabavi, A.; Bertalanffy, H. Role of Delta-Notch signaling in cerebral cavernous malformations. Neurosurg. Rev. 2016, 39, 581–589. [Google Scholar] [CrossRef]
- Quillard, T.; Devallière, J.; Coupel, S.; Charreau, B. Inflammation dysregulates Notch signaling in endothelial cells: Implication of Notch2 and Notch4 to endothelial dysfunction. Biochem. Pharmacol. 2010, 80, 2032–2041. [Google Scholar] [CrossRef]
- Mack, J.J.; Mosqueiro, T.S.; Archer, B.J.; Jones, W.M.; Sunshine, H.; Faas, G.C.; Briot, A.; Aragón, R.L.; Su, T.; Romay, M.C.; et al. NOTCH1 is a mechanosensor in adult arteries. Nat. Commun. 2017, 8, 1620. [Google Scholar] [CrossRef]
- Li, H.; Peng, W.; Jian, W.; Li, Y.; Li, Q.; Li, W.; Xu, Y. ROCK inhibitor fasudil attenuated high glucose-induced MCP-1 and VCAM-1 expression and monocyte-endothelial cell adhesion. Cardiovasc. Diabetol. 2012, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Lisowska, J.; Rödel, C.J.; Manet, S.; Miroshnikova, Y.A.; Boyault, C.; Planus, E.; De Mets, R.; Lee, H.H.; Destaing, O.; Mertani, H.; et al. The CCM1-CCM2 complex controls complementary functions of ROCK1 and ROCK2 that are required for endothelial integrity. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Alexander, R.W.; Medford, R.M. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Investig. 1993, 92, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Khan, B.V.; Harrison, D.G.; Olbrych, M.T.; Alexander, R.W.; Medford, R.M. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox-sensitive transcriptional events in human vascular endothelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 9114–9119. [Google Scholar] [CrossRef] [PubMed]
- Koskimäki, J.; Girard, R.; Li, Y.; Saadat, L.; Zeineddine, H.A.; Lightle, R.; Moore, T.; Lyne, S.; Avner, K.; Shenkar, R.; et al. Comprehensive transcriptome analysis of cerebral cavernous malformation across multiple species and genotypes. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.X.; Liang, L.; Wang, L.; Han, J.T.; Zhu, X.X.; Han, H.; Hu, D.H.; Zhang, P. Inhibition of Notch signaling leads to increased intracellular ROS by up-regulating Nox4 expression in primary HUVECs. Cell Immunol. 2014, 287, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Kovačević, S.; Nestorov, J.; Matić, G.; Elaković, I. Fructose-enriched diet induces inflammation and reduces antioxidative defense in visceral adipose tissue of young female rats. Eur. J. Nutr. 2017, 56, 151–160. [Google Scholar] [CrossRef]
- Xie, X.W. Liquiritigenin attenuates cardiac injury induced by high fructose-feeding through fibrosis and inflammation suppression. Biomed. Pharmacother. 2017, 86, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Cigliano, L.; Spagnuolo, M.S.; Crescenzo, R.; Cancelliere, R.; Iannotta, L.; Mazzoli, A.; Liverini, G.; Iossa, S. Short-Term Fructose Feeding Induces Inflammation and Oxidative Stress in the Hippocampus of Young and Adult Rats. Mol. Neurobiol. 2018, 55, 2869–2883. [Google Scholar] [CrossRef]
- Aquila, G.; Morelli, M.B.; Vieceli Dalla Sega, F.; Fortini, F.; Nigro, P.; Caliceti, C.; Ferracin, M.; Negrini, M.; Pannuti, A.; Bonora, M.; et al. Heart rate reduction with ivabradine in the early phase of atherosclerosis is protective in the endothelium of ApoE-deficient mice. J. Physiol. Pharmacol. 2018, 69, 35–52. [Google Scholar] [CrossRef]
- Hajra, L.; Evans, A.I.; Chen, M.; Hyduk, S.J.; Collins, T.; Cybulsky, M.I. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA 2000, 97, 9052–9057. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.F.; Lenighan, Y.M.; Godson, C.; Roche, H.M. Exploring Coronary Artery Disease GWAs Targets With Functional Links to Immunometabolism. Front. Cardiovasc. Med. 2018, 5, 148. [Google Scholar] [CrossRef]
- Kovacic, S.; Bakran, M. Genetic susceptibility to atherosclerosis. Stroke Res. Treat. 2012, 2012, 362941. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liao, J.K. Rho kinase: An important mediator of atherosclerosis and vascular disease. Curr. Pharm. Des. 2009, 15, 3108–3115. [Google Scholar] [CrossRef] [PubMed]
- Fortini, F.; Vieceli Dalla Sega, F.; Caliceti, C.; Lambertini, E.; Pannuti, A.; Peiffer, D.S.; Balla, C.; Rizzo, P. Estrogen-mediated protection against coronary heart disease: The role of the Notch pathway. J. Steroid Biochem. Mol. Biol. 2019, 189, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Mack, J.J.; Iruela-Arispe, M.L. NOTCH regulation of the endothelial cell phenotype. Curr. Opin. Hematol. 2018, 25, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Boulday, G.; Rudini, N.; Maddaluno, L.; Blécon, A.; Arnould, M.; Gaudric, A.; Chapon, F.; Adams, R.H.; Dejana, E.; Tournier-Lasserve, E. Developmental timing of CCM2 loss influences cerebral cavernous malformations in mice. J. Exp. Med. 2011, 208, 1835–1847. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Drakos, S.G.; Ruiz, O.E.; Smith, A.C.; Gibson, C.C.; Ling, J.; Passi, S.F.; Stratman, A.N.; Sacharidou, A.; Revelo, M.P.; et al. Mutations in 2 distinct genetic pathways result in cerebral cavernous malformations in mice. J. Clin. Investig. 2011, 121, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Do, M.H.; Lee, E.; Oh, M.J.; Kim, Y.; Park, H.Y. High-Glucose or -Fructose Diet Cause Changes of the Gut Microbiota and Metabolic Disorders in Mice without Body Weight Change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieceli Dalla Sega, F.; Mastrocola, R.; Aquila, G.; Fortini, F.; Fornelli, C.; Zotta, A.; Cento, A.S.; Perrelli, A.; Boda, E.; Pannuti, A.; et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. Int. J. Mol. Sci. 2019, 20, 4930. https://doi.org/10.3390/ijms20194930
Vieceli Dalla Sega F, Mastrocola R, Aquila G, Fortini F, Fornelli C, Zotta A, Cento AS, Perrelli A, Boda E, Pannuti A, et al. KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. International Journal of Molecular Sciences. 2019; 20(19):4930. https://doi.org/10.3390/ijms20194930
Chicago/Turabian StyleVieceli Dalla Sega, Francesco, Raffaella Mastrocola, Giorgio Aquila, Francesca Fortini, Claudia Fornelli, Alessia Zotta, Alessia S. Cento, Andrea Perrelli, Enrica Boda, Antonio Pannuti, and et al. 2019. "KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction" International Journal of Molecular Sciences 20, no. 19: 4930. https://doi.org/10.3390/ijms20194930
APA StyleVieceli Dalla Sega, F., Mastrocola, R., Aquila, G., Fortini, F., Fornelli, C., Zotta, A., Cento, A. S., Perrelli, A., Boda, E., Pannuti, A., Marchi, S., Pinton, P., Ferrari, R., Rizzo, P., & Retta, S. F. (2019). KRIT1 Deficiency Promotes Aortic Endothelial Dysfunction. International Journal of Molecular Sciences, 20(19), 4930. https://doi.org/10.3390/ijms20194930