Loss of Apelin Augments Angiotensin II-Induced Cardiac Dysfunction and Pathological Remodeling

 , ,
, ,
Abstract
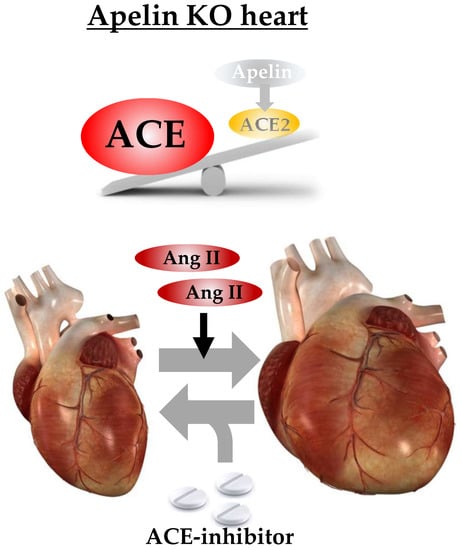
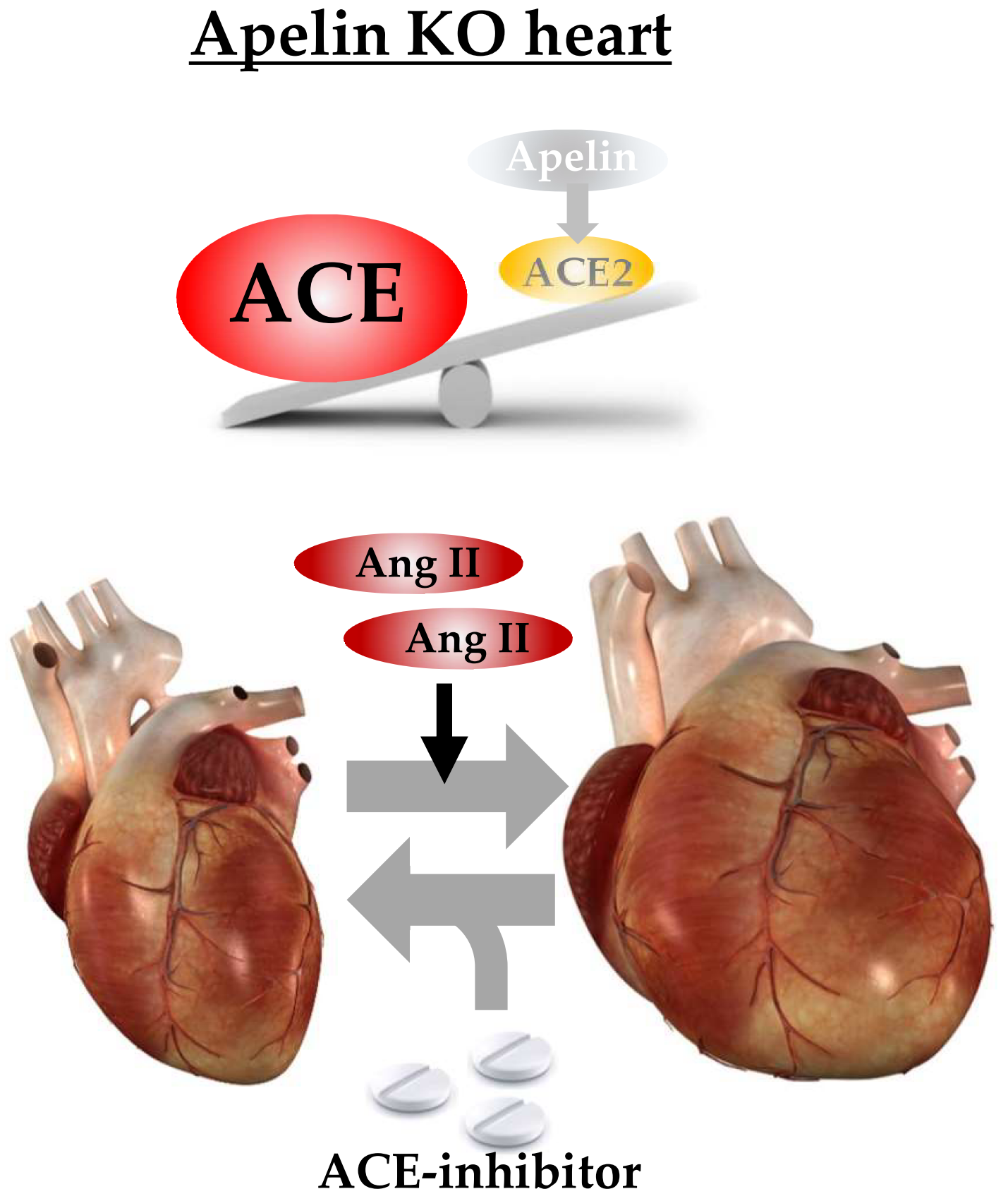
1. Introduction
2. Results
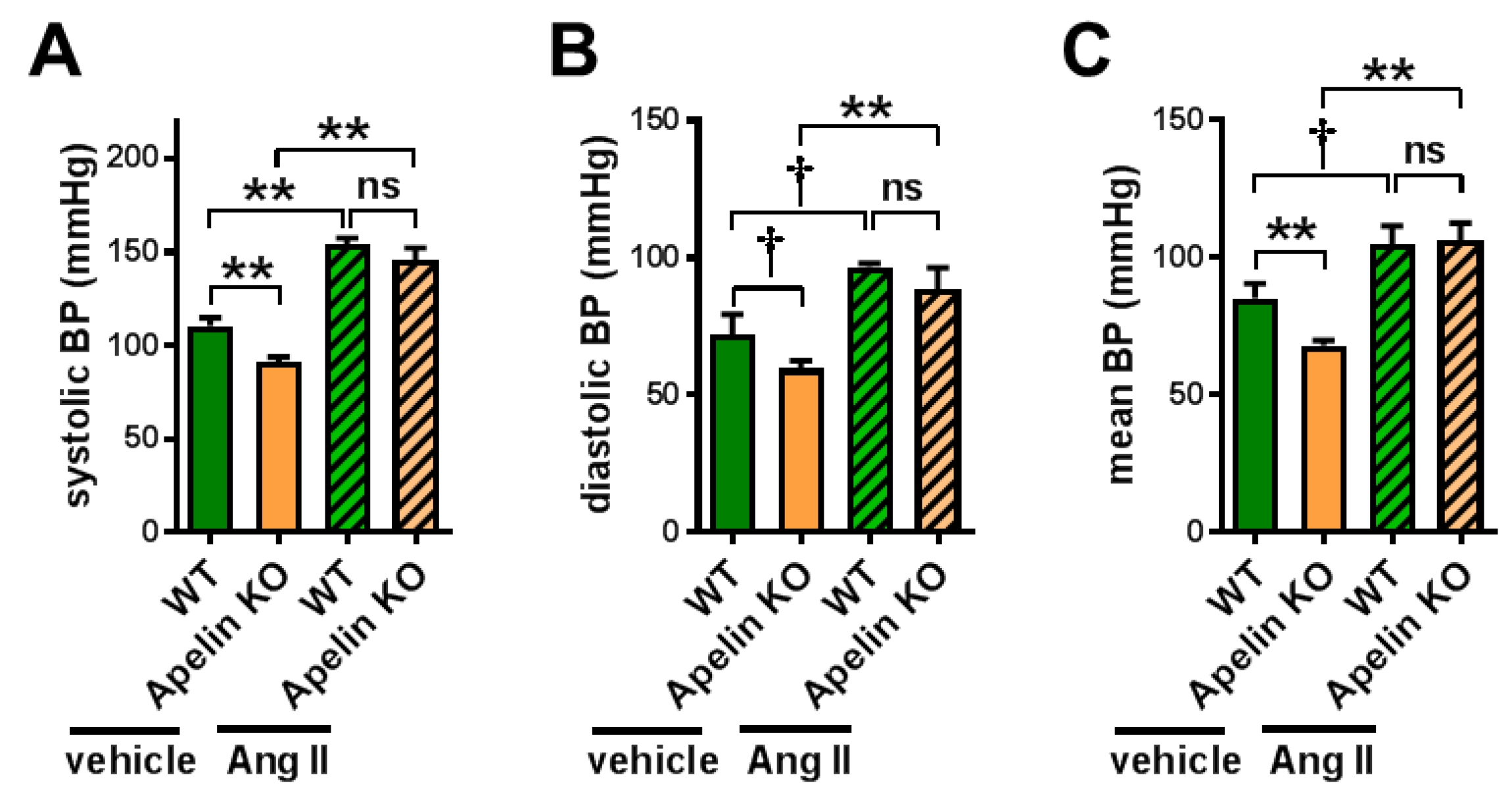
2.1. Apelin Depletion Does Not Affect Ang II-Induced Hypertension in Aged Mice
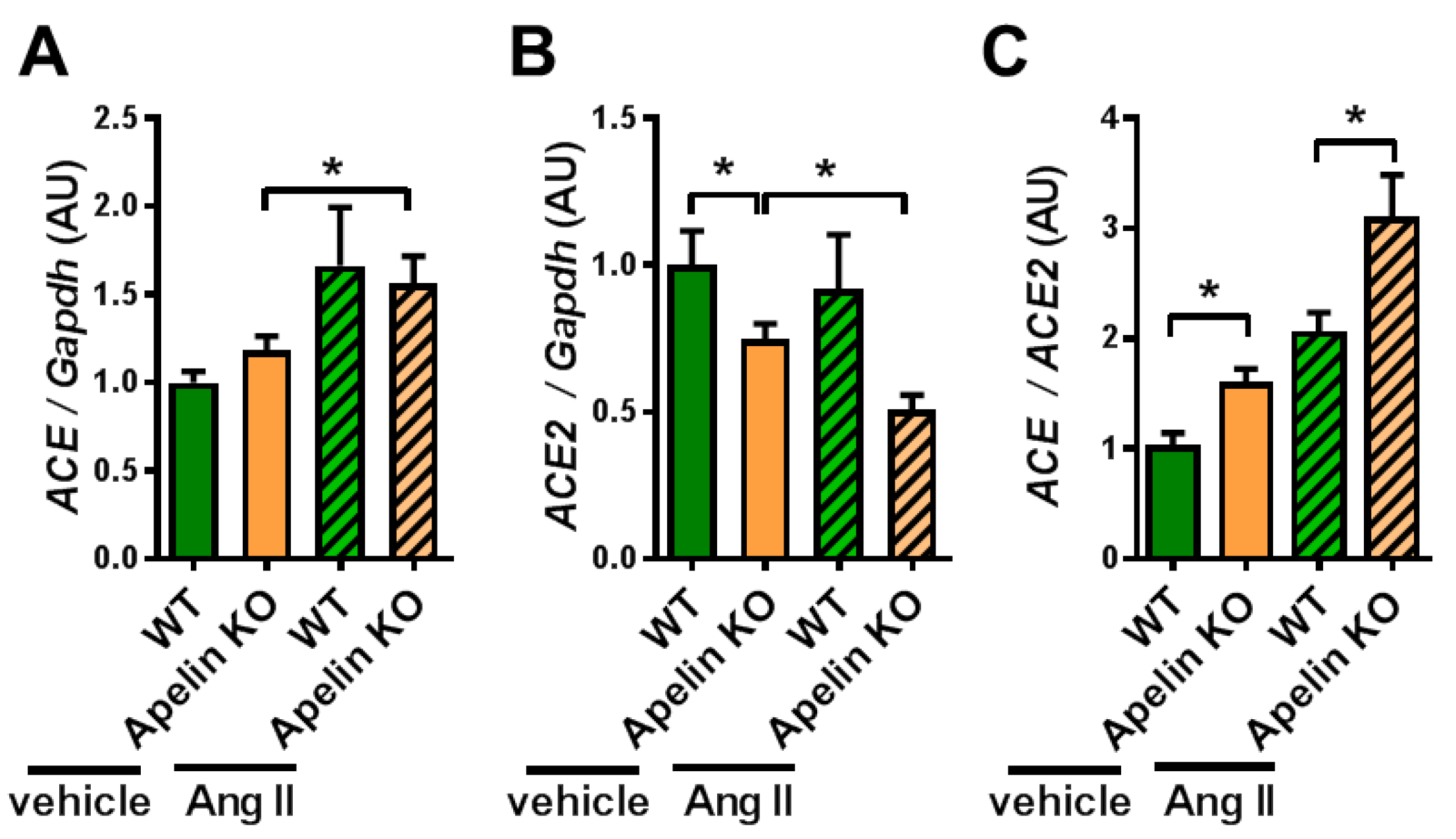
2.2. Ratio of ACE to ACE2 Was Upregulated in the Hearts of Apelin KO Mice
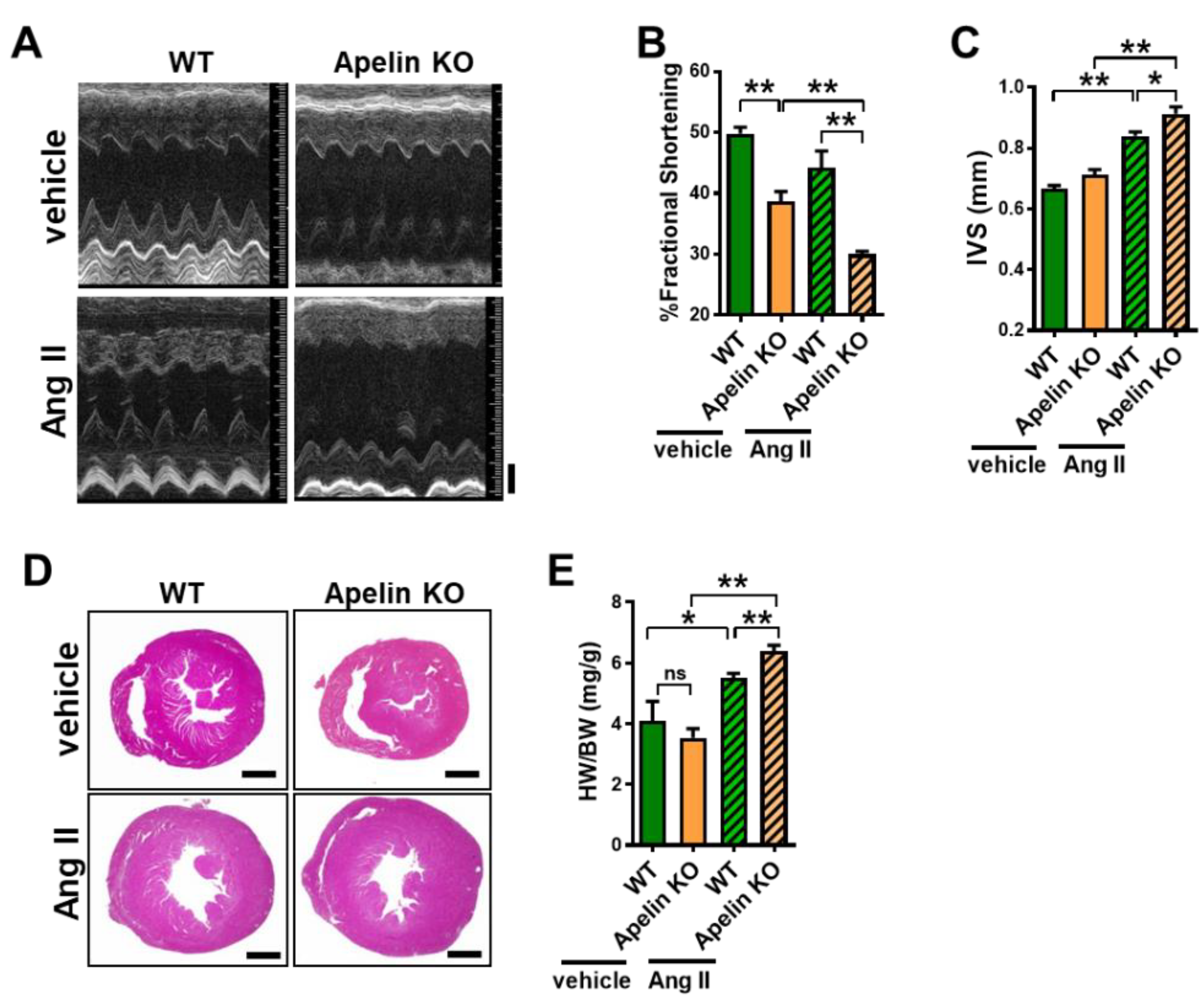
2.3. Ang II Treatment Augments Heart Dysfunction in Aged Apelin KO Mice
2.4. Loss of Apelin Exacerbates Ang II-Induced Cardiac Hypertrophy
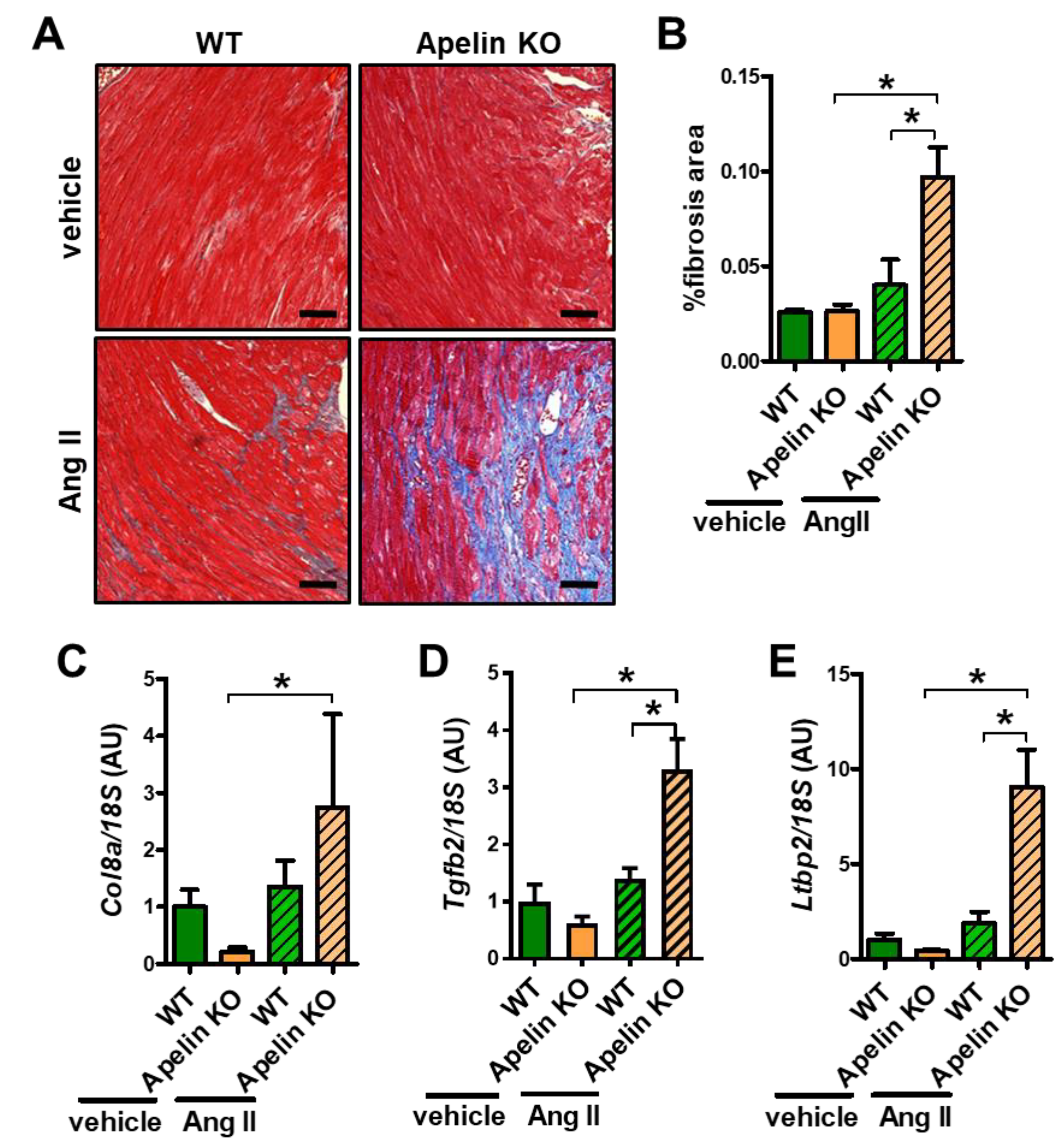
2.5. Ang II Infusion Induces Cardiac Fibrosis in Aged Apelin KO Hearts
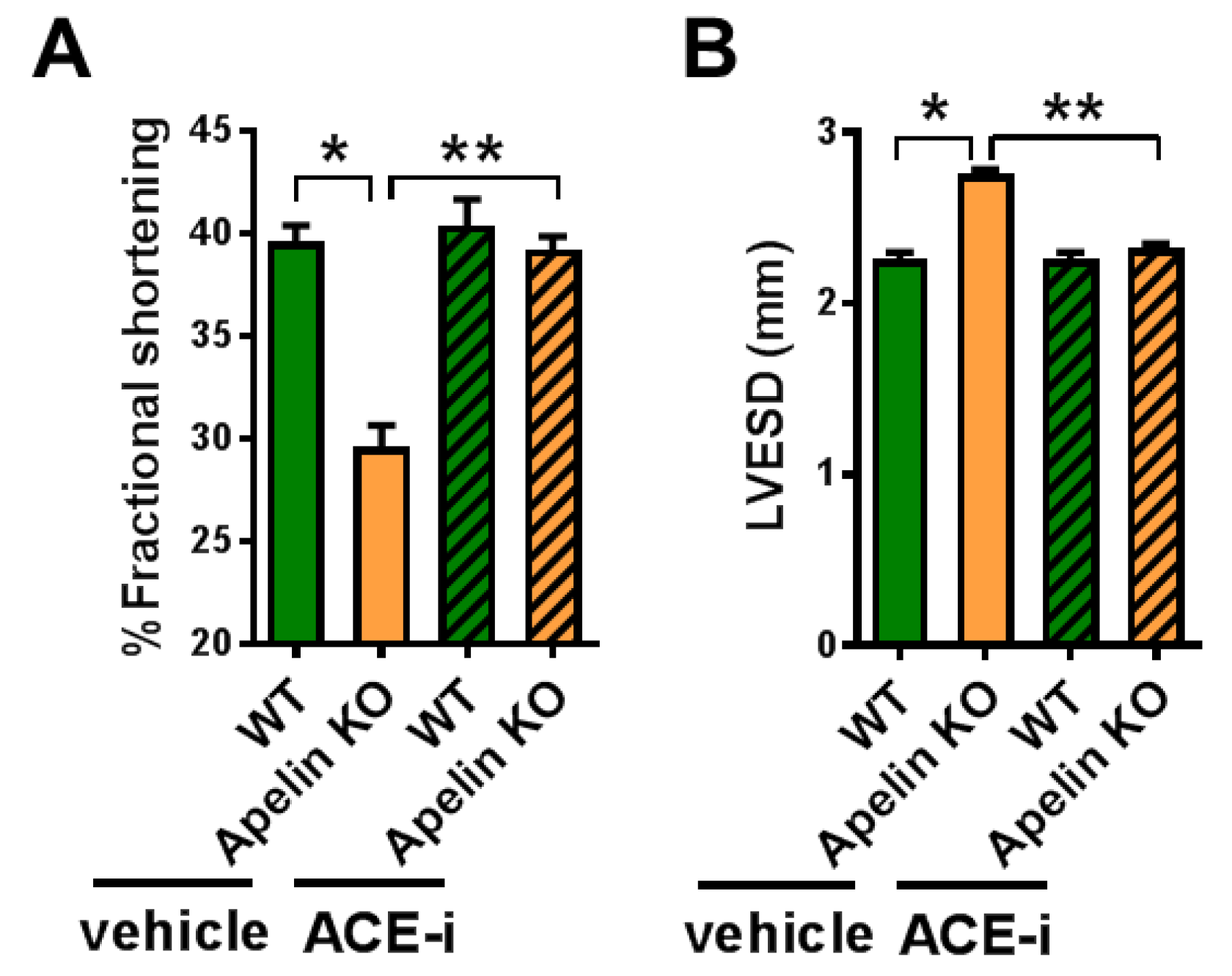
2.6. ACE Inhibitor Improves Impaired Contractility in Aged Apelin KO Hearts
2.7. Apelin Antagonizes Ang II Effects in Primary Cardiomyocytes
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Pharmacological Interventions
4.3. Echocardiography and Blood Pressure Measurements
4.4. LC-MS/MS Analyses for Angiotensin Peptides
4.5. Histology
4.6. Quantitative Real-Time PCR
4.7. Primary Cardiomyocyte Cultures
4.8. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-Converting Enzyme 2 |
| AT1R | Angiotensin Type 1 Receptor |
| RAS | Renin–Angiotensin System |
| TGF-β | Transforming Growth-Factor β |
References
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophy. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Cheng, R.; Nguyen, T.; Fan, T.; Kariyawasam, A.P.; Liu, Y.; Osmond, D.H.; George, S.R.; O’Dowd, B.F. Characterization of apelin, the ligand for the APJ receptor. J. Neurochem. 2000, 74, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Szokodi, I.; Tavi, P.; Földes, G.; Voutilainen-Myllylä, S.; Ilves, M.; Tokola, H.; Pikkarainen, S.; Piuhola, J.; Rysä, J.; Tóth, M.; et al. Apelin, the Novel Endogenous Ligand of the Orphan Receptor APJ, Regulates Cardiac Contractility. Circ. Res. 2002, 91, 434–440. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F.; Heiber, M.; Chan, A.; Heng, H.H.; Tsui, L.C.; Kennedy, J.L.; Shi, X.; Petronis, A.; George, S.R.; Nguyen, T. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene 1993, 136, 355–360. [Google Scholar] [CrossRef]
- Ashley, E.A.; Powers, J.; Chen, M.; Kundu, R.; Finsterbach, T.; Caffarelli, A.; Deng, A.; Eichhorn, J.; Mahajan, R.; Agrawal, R.; et al. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc. Res. 2005, 65, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Japp, A.G.; Cruden, N.L.; Barnes, G.; van Gemeren, N.; Mathews, J.; Adamson, J.; Johnston, N.R.; Denvir, M.A.; Megson, I.L.; Flapan, A.D.; et al. Acute cardiovascular effects of apelin in humans: Potential role in patients with chronic heart failure. Circulation 2010, 121, 1818–1827. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.X.; Pan, C.S.; Zhang, J.; Geng, B.; Zhao, J.; Gerns, H.; Yang, J.; Chang, J.K.; Tang, C.S.; Qi, Y.F. Apelin protects myocardial injury induced by isoproterenol in rats. Regul. Peptides 2006, 133, 147–154. [Google Scholar] [CrossRef]
- Kuba, K.; Zhang, L.; Imai, Y.; Arab, S.; Chen, M.; Maekawa, Y.; Leschnik, M.; Leibbrandt, A.; Markovic, M.; Schwaighofer, J.; et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ. Res. 2007, 101, e32–e42. [Google Scholar] [CrossRef]
- Tao, J.; Zhu, W.; Li, Y.; Xin, P.; Li, J.; Liu, M.; Li, J.; Redington, A.N.; Wei, M. Apelin-13 protects the heart against ischemia-reperfusion injury through inhibition of ER-dependent apoptotic pathways in a time-dependent fashion. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1471–H1486. [Google Scholar] [CrossRef]
- Charo, D.N.; Ho, M.; Fajardo, G.; Kawana, M.; Kundu, R.K.; Sheikh, A.Y.; Finsterbach, T.P.; Leeper, N.J.; Ernst, K.V.; Chen, M.M.; et al. Endogenous regulation of cardiovascular function by apelin-APJ. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1904–H1913. [Google Scholar] [CrossRef]
- Ishida, J.; Hashimoto, T.; Hashimoto, Y.; Nishiwaki, S.; Iguchi, T.; Harada, S.; Sugaya, T.; Matsuzaki, H.; Yamamoto, R.; Shiota, N.; et al. Regulatory roles for APJ, a seven-transmembrane receptor related to angiotensin-type 1 receptor in blood pressure in vivo. J. Biol. Chem. 2004, 279, 26274–26279. [Google Scholar] [CrossRef] [PubMed]
- Scimia, M.C.; Hurtado, C.; Ray, S.; Metzler, S.; Wei, K.; Wang, J.; Woods, C.E.; Purcell, N.H.; Catalucci, D.; Akasaka, T.; et al. APJ acts as a dual receptor in cardiac hypertrophy. Nature 2012, 488, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Ishida, J.; Ishimaru, T.; Mizukami, H.; Hamada, J.; Saito, C.; Fukamizu, A. Lactation is a Risk Factor of Postpartum Heart Failure in Mice with Cardiomyocyte-specific Apelin Receptor (APJ) Overexpression. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [PubMed]
- Capote, L.A.; Mendez Perez, R.; Lymperopoulos, A. GPCR signaling and cardiac function. Eur. J. Pharmacol. 2015, 763, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A. Arrestins in the Cardiovascular System: An Update. Prog. Mol. Biol. Transl. Sci. 2018, 159, 27–57. [Google Scholar] [PubMed]
- Levy, D.; Garrison, R.J.; Savage, D.D.; Kannel, W.B.; Castelli, W.P. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. New Engl. J. Med. 1990, 322, 1561–1566. [Google Scholar] [CrossRef]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Segura, A.M.; Frazier, O.H.; Buja, L.M. Fibrosis and heart failure. Heart Failure Rev. 2014, 19, 173–185. [Google Scholar] [CrossRef]
- Weber, K.T. Targeting pathological remodeling: Concepts of cardioprotection and reparation. Circulation 2000, 102, 1342–1345. [Google Scholar] [CrossRef]
- Schaper, J.; Froede, R.; Hein, S.; Buck, A.; Hashizume, H.; Speiser, B.; Friedl, A.; Bleese, N. Impairment of the myocardial ultrastructure and changes of the cytoskeleton in dilated cardiomyopathy. Circulation 1991, 83, 504–514. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Kassiri, Z.; Patel, M.P.; Chappell, M.; Butany, J.; Backx, P.H.; Tsushima, R.G.; Scholey, J.W.; Khokha, R.; Penninger, J.M. Angiotensin II-mediated oxidative stress and inflammation mediate the age-dependent cardiomyopathy in ACE2 null mice. Cardiovasc. Res. 2007, 75, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ohishi, M.; Katsuya, T.; Ito, N.; Ikushima, M.; Kaibe, M.; Tatara, Y.; Shiota, A.; Sugano, S.; Takeda, S.; et al. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension 2006, 47, 718–726. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Herzenberg, A.M.; Kassiri, Z.; Wong, D.; Reich, H.; Khokha, R.; Crackower, M.A.; Backx, P.H.; Penninger, J.M.; Scholey, J.W. Loss of angiotensin-converting enzyme-2 leads to the late development of angiotensin II-dependent glomerulosclerosis. Am. J. Pathol. 2006, 168, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Bialkowski, K.; Pete, J.; Sheehy, K.; Su, Q.; Johnston, C.; Cooper, M.E.; Thomas, M.C. ACE2 deficiency modifies renoprotection afforded by ACE inhibition in experimental diabetes. Diabetes 2008, 57, 1018–1025. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Therap. 2010, 128, 119–128. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, Y.; Kihara, Y.; Takenaka, H.; Kita, T. Down-regulation of cardiac apelin system in hypertrophied and failing hearts: Possible role of angiotensin II-angiotensin type 1 receptor system. J. Mol. Cell. Cardiol. 2006, 41, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Suzuki, T.; Watanabe, H.; Kadowaki, A.; Fukamizu, A.; Liu, P.P.; Kimura, A.; Ito, H.; Penninger, J.M.; Imai, Y.; et al. Apelin is a positive regulator of ACE2 in failing hearts. J. Clin. Investig. 2013, 123, 5203–5211. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.J.; Ali, Z.A.; Kojima, Y.; Kundu, R.K.; Sheikh, A.Y.; Agrawal, R.; Zheng, L.; Leeper, N.J.; Pearl, N.E.; Patterson, A.J.; et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J. Clin. Investig. 2008, 118, 3343–3354. [Google Scholar] [CrossRef]
- Schreiber, C.A.; Holditch, S.J.; Generous, A.; Ikeda, Y. Sustained ELABELA Gene Therapy in High Salt-Induced Hypertensive Rats. Current Gene Ther. 2017, 16, 349–360. [Google Scholar] [CrossRef]
- Gurley, S.B.; Allred, A.; Le, T.H.; Griffiths, R.; Mao, L.; Philip, N.; Haystead, T.A.; Donoghue, M.; Breitbart, R.E.; Acton, S.L.; et al. Altered blood pressure responses and normal cardiac phenotype in ACE2-null mice. J. Clin. Investig. 2006, 116, 2218–2225. [Google Scholar] [CrossRef] [PubMed]
- Pchejetski, D.; Foussal, C.; Alfarano, C.; Lairez, O.; Calise, D.; Guilbeau-Frugier, C.; Schaak, S.; Seguelas, M.H.; Wanecq, E.; Valet, P.; et al. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur. Heart J. 2012, 33, 2360–2369. [Google Scholar] [CrossRef]
- Lu, L.; Cao, J.; Li, L.; Chen, L. Elabela, a new endogenous ligand of APJ, functions in embryos and adults organisms. Acta Biochim. Biophys. Sin. 2017. [Google Scholar] [CrossRef]
- Parikh, V.N.; Liu, J.; Shang, C.; Woods, C.; Chang, A.C.; Zhao, M.; Charo, D.N.; Grunwald, Z.; Huang, Y.; Seo, K.; et al. Apelin and APJ orchestrate complex tissue-specific control of cardiomyocyte hypertrophy and contractility in the hypertrophy-heart failure transition. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H348–H356. [Google Scholar] [CrossRef]
- Sato, T.; Sato, C.; Kadowaki, A.; Watanabe, H.; Ho, L.; Ishida, J.; Yamaguchi, T.; Kimura, A.; Fukamizu, A.; Penninger, J.M.; et al. ELABELA—APJ axis protects from pressure overload heart failure and Angiotensin II-induced cardiac damage. Cardiovasc. Res. 2017. [Google Scholar] [CrossRef]
- Ho, L.; van Dijk, M. ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science 2017, 357, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Suzuki, T. The CCR4-NOT deadenylase complex controls Atg7-dependent cell death and heart function. Sci. Signal. 2018, 11, eaan3638. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wysocki, J.; Gonzalez-Pacheco, F.R.; Salem, M.; Evora, K.; Garcia-Halpin, L.; Poglitsch, M.; Schuster, M.; Batlle, D. Murine recombinant angiotensin-converting enzyme 2: Effect on angiotensin II-dependent hypertension and distinctive angiotensin-converting enzyme 2 inhibitor characteristics on rodent and human angiotensin-converting enzyme 2. Hypertension 2012, 60, 730–740. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wild Type + Vehicle | Apelin KO + Vehicle | Wild Type + Ang II | Apelin KO + Ang II | |
|---|---|---|---|---|
| N | 4 | 6 | 4 | 6 |
| age (M) | 12 | 12 | 12 | 12 |
| HR (bpm) | 550.28 ± 24.63 | 550.47 ± 50.93 | 577.00 ± 38.03 | 557.67 ± 29.39 |
| FS (%) | 49.83 ± 2.05 | 36.33 ± 6.57 ** | 44.20 ± 5.52 | 31.35 ± 2.20 ## |
| EF (%) | 81.30 ± 2.01 | 65.61 ± 9.08 ** | 75.46 ± 5.92 | 59.44 ± 3.30 ## |
| LVESD (mm) | 2.10 ± 0.20 | 2.76 ± 0.36 ** | 2.19 ± 0.22 | 2.71 ± 0.31 # |
| LVEDD (mm) | 4.18 ± 0.36 | 4.32 ± 0.13 | 3.92 ± 0.45 | 3.95 ± 0.38 |
| IVS (mm) | 0.67 ± 0.02 | 0.71 ± 0.04 | 0.84 ± 0.03 ** | 0.91 ± 0.05 # |
| PWT (mm) | 0.79 ± 0.05 | 0.79 ± 0.04 | 1.00 ± 0.07 ** | 0.95 ± 0.04 |
| Wild Type + Vehicle | Apelin KO + Vehicle | Wild Type + ACE-i | Apelin KO + ACE-i | |
|---|---|---|---|---|
| N | 5 | 5 | 6 | 5 |
| age (M) | 12 | 12 | 12 | 12 |
| HR (bpm) | 476.20 ± 18.00 | 464.40 ± 11.50 | 480.60 ± 11.90 | 455.00 ± 15.90 |
| FS (%) | 39.60 ± 0.81 | 29.60 ± 1.08 ** | 40.40 ± 1.29 | 39.25 ± 0.06 |
| LVESD (mm) | 2.26 ± 0.04 | 2.76 ± 0.02 * | 2.16 ± 0.04 | 2.33 ± 0.02 # |
| LVEDD (mm) | 3.74 ± 0.05 | 3.90 ± 0.05 * | 3.76 ± 0.06 | 3.85 ± 0.06 |
| PWT (mm) | 0.88 ± 0.02 | 0.72 ± 0.02 ** | 0.86 ± 0.02 | 0.83 ± 0.04 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, T.; Kadowaki, A.; Suzuki, T.; Ito, H.; Watanabe, H.; Imai, Y.; Kuba, K. Loss of Apelin Augments Angiotensin II-Induced Cardiac Dysfunction and Pathological Remodeling. Int. J. Mol. Sci. 2019, 20, 239. https://doi.org/10.3390/ijms20020239
Sato T, Kadowaki A, Suzuki T, Ito H, Watanabe H, Imai Y, Kuba K. Loss of Apelin Augments Angiotensin II-Induced Cardiac Dysfunction and Pathological Remodeling. International Journal of Molecular Sciences. 2019; 20(2):239. https://doi.org/10.3390/ijms20020239
Chicago/Turabian StyleSato, Teruki, Ayumi Kadowaki, Takashi Suzuki, Hiroshi Ito, Hiroyuki Watanabe, Yumiko Imai, and Keiji Kuba. 2019. "Loss of Apelin Augments Angiotensin II-Induced Cardiac Dysfunction and Pathological Remodeling" International Journal of Molecular Sciences 20, no. 2: 239. https://doi.org/10.3390/ijms20020239
APA StyleSato, T., Kadowaki, A., Suzuki, T., Ito, H., Watanabe, H., Imai, Y., & Kuba, K. (2019). Loss of Apelin Augments Angiotensin II-Induced Cardiac Dysfunction and Pathological Remodeling. International Journal of Molecular Sciences, 20(2), 239. https://doi.org/10.3390/ijms20020239